- LOGIN
- MemberShip
- 2026-06-10 16:16:04

- Policy
- Multi-listing rule hits non-innovative companies harder
- by Jung, Heung-Jun May 19, 2026 11:08am
- The ‘multi-product listing management’ rule, introduced to prevent excessive proliferation of generics, will operate far more harshly against non-innovative companies.While innovative and quasi-innovative companies will receive a three-year grace period, general companies will face immediate price cuts to 30.6%-38.5%, depending on whether they pass bioequivalence testing, leading to a significant widening of the price gap.According to industry sources on the 18th, the impact of the multi-product listing penalty system, newly introduced starting this August, will differ dramatically depending on a company’s innovation classification.Impact of the multi-product listing penalty systemThe Ministry of Health and Welfare recently issued an administrative notice revising the “Standards for Drug Price Determination and Adjustment,” newly introducing a provision applying 85% of the calculated price once the total number of identical formulations reaches 14 or more. Drugs eligible for additional premiums will also be subject to the 85% rule once the premium period ends.For innovative and quasi-innovative companies, the drug price premium period is 1+3 years if domestic manufacturing conditions are met. In other words, a 3-year grace period applies to the multi-product listing penalty as well.In contrast, general companies face an immediate reduction from the standard calculation rate of 45% to 38.25% once the number of listed products exceeds 14. If they failed to conduct their own bioequivalence testing, the price is reduced by an additional 20%, falling to 30.6%.For example, if a generic drug with an original product price of KRW 1,000 reaches 14 or more listed generics, an innovative company would maintain a price of KRW 600 won, reflecting a 60% premium, for four years before eventually dropping to KRW 382.5. A quasi-innovative company would maintain KRW 500 under a 50% premium for four years before dropping to KRW 382.5.However, the generic drug of a general company would sell at KRW 450 for only one year under the standard calculation rate before falling to KRW 382.5.This means that while innovative and quasi-innovative companies maintain prices of KRW 500–600 for three years by meeting domestic manufacturing requirements, general companies immediately face deteriorating profitability at KRW 382.5.If an ordinary company additionally failed to meet the bioequivalence testing requirement, the price would fall to KRw 306, creating nearly a twofold price gap compared with innovative companies.Ultimately, this structure ensures that the penalty for listing multiple products is applied more strictly to general companies, and a sales gap with innovative and quasi-innovative companies is inevitable during the grace period for the price adjustment.In effect, general companies effectively lose the advantage of product listing after the 14th generic entrant, subject to the multi-product pricing rule, enters the list.

- Opinion
- "Eliminating generic pharma's shared bio-equivalence·illegal CSO"
- by Lee, Jeong-Hwan May 19, 2026 11:08am
- Won-jun Jo, Chief Representative for Health and Welfare of the Democratic Party's Policy Committee"The goal of the Democratic Party’s drug pricing system reform is not simply to reduce the National Health Insurance budget by cutting generic prices. The goal is to eliminate the space for paper-company pharmaceutical companies and free-riders that parasitize the pharmaceutical ecosystem, thereby establishing an industry order in which companies that properly manufacture and invest in novel drugs and supply unstable medicines are dramatically rewarded. This is also a principle we have firmly held in our general and presidential election pledges. Now that the reform plan has been finalized, the ruling party will work with the government to complete the remaining puzzle pieces of the drug pricing system through the abolition of the generic '1+3' shared bioequivalence system and follow-up measures to regulate unsound CSOs (pharmaceutical sales marketing agencies)."As the government finalized the broad framework and key details of the drug pricing system reform plan, the Democratic Party of Korea drew attention by expressing a strong determination to accelerate policies aimed at eliminating illegal rebates through the abolition of the generic 1+3 shared bioequivalence test system and the regulation of poor CSOs to complete the mission of "pharmaceutical and biotech industry innovation."By lowering the generic drug price calculation rate from 53.55% to 45% and strengthening selective and differential incentives compared to the past for innovative pharmaceutical companies, semi-innovative pharmaceutical companies, and companies contributing to supply-unstable medicines, the party intends to thoroughly exclude name-only pharmaceutical companies from the market that do not align with the values of pharmaceutical industry development, healthy employment and job creation, and the establishment of a sound medicine distribution structure.On the 17th, Won-jun Jo, Chief Representative for Health and Welfare of the Democratic Party's Policy Committee, met with DailyPharm and expressed, "Starting with the Ministry of Health and Welfare's drug pricing system reform plan, we are discussing follow-up policies that can send a clear and unambiguous message to the pharmaceutical and biotech industry, encompassing both novel drugs and generics, as well as the CSO industry."Jo evaluated that the Ministry of Health and Welfare's drug pricing system reform plan, which passed the Health Insurance Policy Review Committee and is set for implementation this year, deleted remaining inefficiencies and irrationalities while presenting a future vision for South Korea's pharmaceutical industry to pursue.The aim is to smartly restructure the reward system for pharmaceutical companies that contribute to novel drug research and development (R&D), the stable supply of essential medicines, and the growth of the national pharmaceutical industry, so that the nation and its citizens achieve a tangible level of practicality they can directly perceive."Novel Drug National Health Insurance Reimbursement, from an 'Admissions Quota System' to a 'Graduation Quota System'Jo explained that the discussions and designs for the drug pricing system reform with the Ministry of Health and Welfare (MOHW) focused primarily on significantly narrowing the gap between the public and novel drugs under limited health insurance budget conditions, and satisfying the social demand to resolve the issue of essential medicines that frequently go out of stock due to supply instability.Because the expansion of public access to novel drugs was incorporated into the reform plan, some civil and patient advocacy groups raised concerns that it might grant excessive privileges to global pharmaceutical companies focused on novel drugs, a perspective Jo believes should also be adequately acknowledged.In particular, Jo emphasized that the speed of implementing health insurance reimbursement to novel drugs is not the only factor that is increasing. He stated that, following the rapid reimbursement of a novel drug, a follow-up policy based on a mechanism to remove it from reimbursement immediately will be implemented if its drug efficacy cannot be proven through RWD.Jo said, "Until now, the barrier to entering novel drug reimbursement was high, and once a drug received reimbursement, it was a structure where reimbursement was continuously recognized thereafter. There were criticisms that this system was actually more irrational," and explained. "A judgment was made that it is more efficient to change the reimbursement barriers and criteria to be relatively flexible, but transition to a system where reimbursement is deleted if real-world prescription efficacy data is not clear during post-evaluations."He added, "We will proceed with follow-up work to prepare policies capable of determining reimbursement ejection based on real-world data. This is a package that the drug pricing system reform plan must accompany. To use a simple analogy to the college admission system, we are transitioning novel drug reimbursement from an admissions quota system to a graduation quota system. It changes to a graduation quota system where reimbursement cannot be maintained unless clear evidence of drug efficacy is proven after entering reimbursement.""Free-riding pharmaceutical companies must be sorted out for good generics to distribute"Regarding the significance of the generic drug pricing system reform, Jo summarized it as meaning "there are no drug prices to give to free-riding pharmaceutical companies.""Can a company that does not perform its own bioequivalence testing, its own clinical trials, or even its own direct manufacturing be called a pharmaceutical company?" Jo asked and assessed that "If you give the same drug price to a consignment pharmaceutical company simply because it has the same ingredient, that company has no reason to invest in personnel or spend money on infrastructure. Consequently, they become entirely consumed by maximizing generic sales competitiveness, which deteriorates into a structure that inevitably links to illegal rebates."Jo noted, "The major meaning of the generic reform plan is not about cutting drug prices, but rather about rewarding only those pharmaceutical companies that manufacture proper generics with a proper drug price. We believed a policy response was needed to address whether it is right to continue embracing consignment generics within the health insurance system. Therefore, we adjusted and newly established criteria for innovative and semi-innovative pharmaceutical companies, and embedded regulations in the reform plan that can yield benefits for companies contributing to supply-unstable medicines."Regarding the reduction of the generic calculation rate to 45%, Jo evaluated, "Although it may not be completely satisfactory to either the government or the pharmaceutical industry, an agreement was reached at a level that cannot be viewed as fatal to either side at the same time.""The pharmaceutical industry demanded 48% as a baseline margin, while the MOHW discussed the low 40% range. Looking only at the surface, it is the product of a social consensus, and political circles adjust aspects," he said. "It was also determined at a median level between the calculation values of Japan and France, which were referenced during the system design. This is why the pharmaceutical industry, which had major anxieties, was able to say after the system was finalized that it was difficult. They must endure a portion of it."Jo stated that the "graduation quota system" for novel drug reimbursement, the abolition of the 1+3 consignment generic system, and the eradication of illegal CSO rebates are the path the domestic pharmaceutical industry must take."Abolition of the '1+3' shared bioequivalence system is the goal"Jo asserted with confidence that the generic consignment bioequivalence system must be completely abolished so that paper-company pharmaceutical companies that free-ride on pharmaceutical industry development and the national health insurance budget disappear, establishing a pharmaceutical industry environment where only genuine pharmaceutical companies receive justified rewards.Jo's opinion that the current method, which allows three shared consignment pharmaceutical companies for every one pharmaceutical company performing a generic bioequivalence test, is a contradictory policy that stands at the opposite pole of the reformed drug pricing system.Jo announced that he will take the necessary legislative and administrative steps, alongside the MOHW and the Ministry of Food and Drug Safety, to implement a system that allows only a single generic per original medicine. This policy is expected to exert a significant impact on the overall structure of the domestic pharmaceutical industry, increasing the need to focus on future National Assembly legislation and government administrative movements."Currently, the 1+3 consignment bioequivalence generic system is permitted, but the rationale for why identical health insurance reimbursement drug prices should be given to three consignment generic items cannot be explained by any logic. It completely runs counter to the philosophy of the reformed drug pricing system," Jo emphasized. "In the past, it carried the meaning of partially cleaning up the market as a transitional phase, shifting from permitting unlimited bioequivalence tests to restricting it to 1+3, but now, the biggest characteristic of the reformed drug pricing system is that it will not give drug prices to pharmaceutical companies that free-ride by permitting generic consignment bioequivalence and manufacturing."Jo pointed out, "The consignment bioequivalence system has reached the point where it must be abolished. Previously, the MFDS also stated its position when announcing the 1+3 system that it was a temporary and provisional allowance. Some argue that the abolition of 1+3 will lead to job reductions. It is questionable what kind of industrial or national production consignment generic companies, which have a large proportion of paper companies, induce, or what employment creation effects they demonstrate.""Rebates exploiting rogue CSOs are evolving...We will eliminate these cases"Jo cited the elimination of CSO rebates as one of the critical follow-up measures that must be implemented after the drug pricing reform.Jo stated that, alongside the ejection of free-riding consignment pharmaceutical companies, regulations must be placed on malpractice in which some pharmaceutical companies exploit CSOs to continue illegal rebate sales operations to secure unfair profits, thereby amplifying the effects of the reformed drug pricing system."For good generics made by real pharmaceutical companies to be properly distributed in the market and for citizens to take their medications, the next targets for restructuring are unsound CSOs and the pharmaceutical companies that exploit them," Jo pointed out. "Like an open secret, some pharmaceutical companies select and operate with CSOs as indirect actors to diversify the risks and responsibilities of illegal rebates. These CSOs operate under a subcontracting and further re-subcontracting structure, ultimately making it ambiguous as to who bears the final liability for the rebate activity."Accordingly, he introduced, "Recently, some hospitals and directors have been disrupting the order of the sound medicine market by operating family CSOs as a method of evasion for family business succession to secure illegal profits. We are contemplating measures to strengthen CSO compliance alongside the Ministry of Health and Welfare. One of the measures under consideration is making it fundamentally impossible for a pharmaceutical company to cut off its relationship with a CSO to evade responsibility when a rebate is detected."In conclusion, Jo said, "We will strengthen regulations on malpractice CSOs through a joint penalty system for pharmaceutical companies and CSOs to eliminate problems where CSOs make it impossible to trace the origin of a rebate by subcontracting and re-subcontracting, and pharmaceutical companies cut relationships and deflect responsibility by blaming the CSO. Legislation that clarifies the mutual chain of responsibility between a pharmaceutical company and its contracted CSO will follow to support the success of the drug pricing reform plan."
- InterView
- [Desk's View] The 16-year shackle around K-toxin must be broken
- by Lee, Seok-Jun May 19, 2026 11:08am
- The declassification of botulinum toxin as a National Core Technology is under discussion. This time, the atmosphere is noticeably different. It has been reported that the Biotechnology Expert Committee under the Ministry of Trade, Industry and Energy (MOTIE) has reached internal consensus in favor of declassification. All that remains now is a conclusion.According to MOTIE and industry sources, the Biotechnology Committee, operating under the Industrial Technology Protection Committee, is currently reviewing whether to remove botulinum toxin product manufacturing technology and its strains from the National Core Technologies designation list. MOTIE states that this is part of a routine, periodic review process to reassess 79 national core technologies across sectors such as semiconductors, displays, and biotechnology.However, the market does not view this as a mere routine review. This is because a regulatory controversy that has persisted for 16 years is shifting direction for the first time.In fact, sources inside and outside the industry observe that the Biotechnology Committee has reached consensus primarily on the declassification view. Assessments also indicate that the atmosphere shifted noticeably after MOTIE recently replaced several long-tenured, consecutively reappointed committee members.This change is significant. Previously, the controversy surrounding the national core technology designation for botulinum toxin focused on the issue of specific committee members serving long, consecutive terms. During last year's National Assembly audit, critics pointed out the expert committee's closed nature and its structural reliance on specific individuals. MOTIE's personnel overhaul aligns directly with this trend, implying that the newly appointed expert committee members for this year have converged on the opinion of lifting the designation. The core issue is simple. The current discussion is not about whether to unlock a single technology. It is about whether the outdated regulatory framework can be normalized.Meanwhile, the domestic botulinum toxin industry has long argued that the National Core Technology designation constitutes excessive, duplicate regulation. Their view is that because management systems are already functioning under the Pharmaceutical Affairs Act, the Infectious Disease Control and Prevention Act, and the Foreign Trade Act, layering National Core Technology regulations on top has only inflated administrative burdens.Critics have pointed out that South Korea is the only country to designate botulinum toxin as a National Core Technology, even though it is recognized globally as a generic manufacturing technology. Furthermore, the strains are publicly available on global genetic information networks, and a significant number of domestic companies utilize overseas strains. Despite this, additional reviews were repeatedly required in South Korea for every export, technical cooperation, and licensing process. Ultimately, it was a structure where administration outpaced industrial development.The problem lies in the outcome. The global botulinum toxin market has reorganized around American and European companies. Even though domestic companies possess manufacturing competitiveness, they have found themselves tied down by various administrative procedures. Uncertainty has also repeatedly plagued overseas market expansion and technology transfer negotiations.The National Core Technology system itself cannot be discredited. However, regulations must keep pace with reality. If the restrictive effects on industry outweigh the actual benefits of protection, the policy must be amended.Above all, these discussions must not end in another round of dragging time. In the past, whenever the possibility of declassification was raised, postponing a conclusion was a recurring pattern. In the meantime, market uncertainty only intensified.Now, the situation is different. A shift in the committee's internal current has been detected, and MOTIE has signaled a personnel overhaul. Now, only the final judgment remains.If the shackles that have bound K-toxin for 16 years cannot be broken this time either, the market will inevitably ask once again. For whose benefit is the South Korean biotech industry being regulated?The ball is now in the Industrial Technology Protection Committee's court. Conclusion awaits.

- Company
- Blincyto enters pricing negotiations as consolidation therapy
- by Eo, Yun-Ho May 19, 2026 11:08am
- The blood cancer treatment ‘Blincyto’ has entered the final stage of the process to expand its health insurance coverage in Korea.According to industry sources, Amgen Korea recently began drug price negotiations with the National Health Insurance Service regarding reimbursement for Blincyto (blinatumomab) as consolidation therapy for precursor B-cell acute lymphoblastic leukemia (ALL).The indication, which received expanded approval in Korea in February 2025, previously passed the Health Insurance Review and Assessment Service’s Drug Reimbursement Evaluation Committee in April.Patients with Philadelphia chromosome-negative (Ph-) precursor B-cell ALL frequently experience relapse even after achieving minimal residual disease (MRD)-negative status through conventional chemotherapy-based induction therapy, and continue to face challenges with long-term survival even after hematopoietic stem cell transplantation, indicating a significant unmet medical need.Blincyto’s consolidation therapy indication demonstrated efficacy through the E1910, AALL1731, AALL1331, and 20120215 studies.In the E1910 study, which compared chemotherapy alone versus alternating Blincyto and chemotherapy as post-induction consolidation therapy in adult precursor B-cell ALL patients, the 3-year overall survival (OS) rate among MRD-negative patients was 85% in the Blincyto-plus-chemotherapy alternating group, compared to 68% in the chemotherapy-alone group.Compared with chemotherapy alone, the Blincyto-plus-chemotherapy alternating group showed a 59% reduction in risk of death over a median follow-up period of 43 months.In addition, the 3-year recurrence-free survival (RFS) rate was 80% in the Blincyto-plus-chemotherapy alternating group versus 64% in the chemotherapy-alone group, representing a 47% reduction in the risk of recurrence or death over a median follow-up of 43 months.Furthermore, results from the AALL1731 study involving MRD-negative pediatric precursor B-cell ALL patients in the National Cancer Institute (NCI) standard-risk (SR) category at average or high risk of relapse showed that the estimated 3-year disease-free survival rate at a median follow-up of 2.5 years was 96.0% in the Blincyto-plus-chemotherapy alternating group, s a significant improvement compared to 87.9% in the chemotherapy-alone group.Meanwhile, the ‘2024 National Comprehensive Cancer Network (NCCN) Guidelines’ recommend a regimen that includes Blincyto as first-line consolidation therapy.

- Company
- Eli Lilly Korea's CEO transition…Seiya Komatsu expected to be named
- by Eo, Yun-Ho May 18, 2026 09:11am
- The CEO of Eli Lilly Korea is expected to be replaced.According to industry sources, Eli Lilly has appointed Seiya Komatsu as the new CEO of its Korean affiliate.The appointment comes as the term of the current president, John Bickel, is set to end.As of the end of this coming July, John Bickel, who took office in August 2024, is scheduled to be promoted to a position at Eli Lilly's global headquarters.The newly appointed CEO, Seiya Komatsu, is an industry expert who joined Eli Lilly Japan as a sales representative in 2012 and has acquired diverse experience across various roles, including brand marketing manager, business transformation consultant at global headquarters, and sales manager for the Texas region in the United States. Currently, Komatsu serves as Vice President and Head of the Neuroscience Business Unit at the Japanese affiliate.Meanwhile, Eli Lilly supplies pharmaceuticals across various therapeutic areas, including oncology and autoimmune diseases, as well as its obesity treatment, 'Mounjaro.' Notably, Mounjaro recorded global sales of USD 8.7 billion (approximately KRW 12.7 trillion) in the first quarter of this year, surpassing MSD's 'Keytruda,' ranking No.1 in global pharmaceutical sales performance.
- Policy
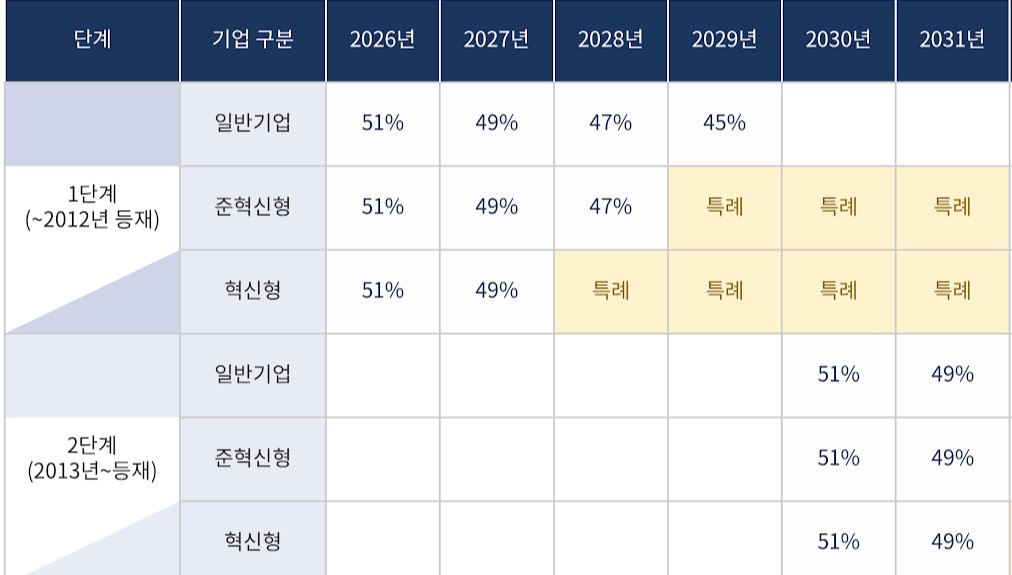
- Price cuts on existing drugs divided into two phases
- by Jung, Heung-Jun May 18, 2026 09:11am
- As the government proceeds with planned pharmaceutical pricing system reforms, follow-up discussions are expected regarding specific classification criteria for price reductions of pre-listed drugs.Key issues include how to distinguish between first-phase and second-phase price reductions for two- and three-drug combination products, as well as how and when later bioequivalence testing should be reflected in drug price reductions.According to industry sources on the 15th, specific criteria for price reductions on pre-listed drugs have not yet been finalized. Discussions on price reductions for pre-listed drugs were not properly addressed during the working-level consultative body meeting between the government and the pharmaceutical industry held in late April.Previously, the Health Insurance Review and Assessment Service (HIRA) decided to divide drugs into two groups based on their listing date in 2012. Price reductions for ingredients listed before 2012 will begin immediately within this year, while reductions for Phase 2 drugs, listed after 2012, will begin in 2030.The timing of implementation differs depending on whether a product is classified into the first or second phase of existing drug price cutsHowever, opinions remain divided regarding the classification of combination products. If the individual ingredients making up a combination drug belong to both Phase 1 and Phase 2 groups, authorities must determine at which timing the combination product itself will face price reductions.The government has stated that if even one ingredient in a combination product was listed before 2012, the combination drug would be classified as a Phase 1 drug.However, there are many issues that require consultation with the industry, such as how to handle cases where even a single ingredient retains a patent or has PMS remaining.Another key issue is how to reduce prices for drugs that failed to meet required standards due to a lack of bioequivalence testing. The reduction rate under the differentiated standards system has been increased from 15% to 20%.For example, if a product price was lowered to 45% and bioequivalence testing was not conducted, it would fall further to 36%. If a rate of 49% is applied, being a product from an innovative pharmaceutical company, it would become 39.2%, while applying a quasi-innovative rate of 47% would drop the price to 37.6%.Since price reductions for pre-listed drugs are implemented over a 10-year period, including a grace period, it is also important to determine how to reflect the results if bioequivalence testing is conducted during that period.In particular, since Phase 2 drugs will only begin facing reductions in 2030, some pharmaceutical companies may attempt to protect reduction rates by conducting bioequivalence studies.Accordingly, industry players are expected to argue that satisfying bioequivalence requirements by the final year converging toward the calculation rate should still qualify as meeting standard requirements.
- Policy
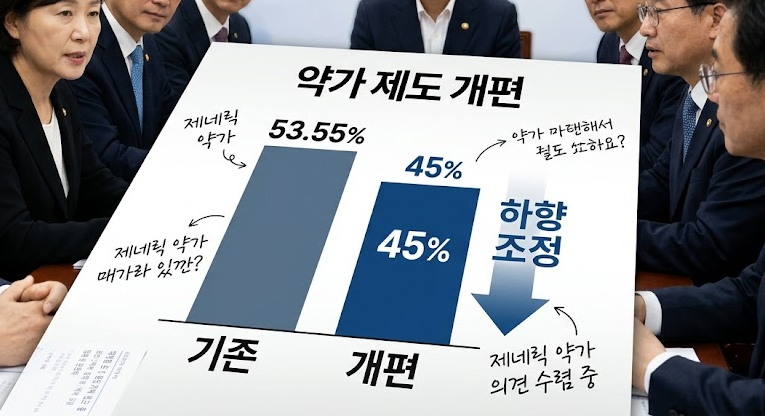
- Calculation rate for generic drug prices set at 45%
- by Lee, Jeong-Hwan May 18, 2026 09:11am
- The government has announced that a drug pricing reform plan, which cuts the drug price calculation rate for currently listed generics from 53.55% to 45%, will take effect on August 1st.The reform plan also includes improvements regarding the management of multiple-listed items, drug price calculations for transfers and acquisitions, support criteria for drugs subjected to market withdrawal, and criteria for semi-innovative pharmaceutical companies.On the 14th, the Ministry of Health and Welfare (MOHW, Minister Jung Eun-kyeong) issued a public notice for the partial amendment to the "Criteria for determination and adjustment of drugs." The ministry plans to finalize the amendment after gathering public feedback by July 13th.The implementation date for the drug pricing reform specified in the amendment notice is August 1st. The government announced that it will gather opinions until July 13th.Generic drug price calculation rate 45%....products failing to meet requirements will be priced below 36%First, the drug price calculation rate for currently listed generics will be reduced from 53.55% to 45%. The calculation rate applied to generics that fail to meet the baseline requirements will also be reduced from 85% to 80%.The baseline requirements for the drug price include whether the company conducted an independent bioequivalence test and whether it used registered drug master file (DMF) ingredients.Generics that meet all baseline requirements will be priced at 45%, those meeting some requirements will be priced at 36%, and products failing to meet any requirements will be priced at 29%.In the case of tiered pricing, the price will be cut once the number of listed items with the same formulation exceeds 13. This is a tighter restriction compared to the current threshold of 20 or more items.If the sum of the newly applied product and the number of currently listed items with the same formulation exceeds 14, the upper limit price will be fixed at 85% of the calculated amount once the price premium period ends.Innovative pharmaceutical companies, semi-innovative pharmaceutical companies, and supply-stabilizing leading pharmaceutical firms will receive preferential drug pricing. Among drugs that meet all baseline requirements, items from innovative pharmaceutical companies will receive a 60% price premium. The premium rate for items from semi-innovative pharmaceutical companies or supply-stabilizing leading pharmaceutical companies will be 50%.Definitions for semi-innovative pharmaceutical companies and supply-stabilizing leading pharmaceutical companies were also established. A supply-stabilizing leading pharmaceutical company is defined as a firm whose ratio of low-profit prevention support drugs, or the ratio of billing amounts among its listed drugs, is 20% or higher.Regarding transfers and acquisitions, the government decided to restrict the succession of existing upper limit prices for items involving a change in a manufacturer's status, excluding inheritance or mergers. Even if a generic item that maintains a high drug price is acquired, the recalculated drug price will be applied from the time of the transfer and acquisition.This regulation is designed to block back-door strategies to evade drug price cuts by purchasing items that maintain high prices.Support for drugs facing production discontinuation will be strengthened. The designation criteria for these drugs are KRW 578 for oral medications, KRW 44 per minimum unit for oral liquids, KRW 3,080 for external preparations, and KRW 5,783 for injections.A new premium clause was also created for pharmaceutical companies that have contributed to supply stabilization. The premium evaluation items include the track record of stable supply fulfillment, national essential medicines, single-listed medicines, low-priced medicines, the use of domestically produced raw ingredients, an annual billing amount of less than KRW 500 million in the previous year, treatments for statutory infectious diseases, and infectious disease crises or urgent supply shortage situations.Price-volume linkage system officiated…implemented on April 1st and October 1st of each yearThe timing of drug price cuts resulting from the price-volume linkage system and the expansion of the scope of use has been unified. The amendment specified that the ex officio adjustment of the upper limit price of drugs will be implemented on April 1st and October 1st of each year, unless there are special circumstances.In addition, a basis was established for pharmaceutical companies to refund the increased expenditure on health insurance incurred during the implementation grace period to the National Health Insurance Service if a drug price adjustment is issued at a time other than the regular implementation date.Meanwhile, the MOHW plans to implement the announced reform plan on August 1st. Regulations related to the regularization of the price-volume linkage will apply starting in January of next year (2027). The first regular drug price adjustment will take effect on April 1st, 2027.

- Product
- Regulatory sandbox for OTC drug vending machines revisited
- by Kim JiEun May 18, 2026 09:11am
- Discussions surrounding a regulatory sandbox pilot exemption for “smart vending machines for over-the-counter essential medicines,” which has remained unresolved for years, have recently resumed. With the expert committee under the Ministry of Trade, Industry and Energy’s Regulatory Exemption Deliberation Committee convening again after about 2-3 years, debate over permitting unmanned sales of OTC essential medicines appears to be reigniting.In particular, it is reported that a heated debate ensued during this meeting, with the industry strongly advocating for the pilot exemption by emphasizing the need for installation in medically underserved areas and technological advancements, while the Korean Pharmaceutical Association and the Ministry of Health and Welfare clearly stated their opposition, as they have in the past.According to multiple sources on the 15th, the Ministry of Trade, Industry, and Energy held the “5th Expert Committee Meeting of the Regulatory Rationalization Committee” at the end of last month to discuss a proposal for a pilot exemption regarding smart vending machines for over-the-counter medicines.This marked the first reopening of discussions on the issue in approximately two to three years after deliberations had effectively been suspended. It was reported that attendees included representatives from the Korean Pharmaceutical Association, the Ministry of Health and Welfare, the Ministry of Trade, Industry and Energy, the applicant company Urban Sharing Economy, as well as legal and academic experts.The core of this proposal is to allow the sale of over-the-counter medicines through unmanned smart vending machines. Previously, the company had applied for a regulatory sandbox, proposing a smart vending machine model that integrates biometric authentication and remote control systems based on 13 over-the-counter medication items currently sold at convenience stores.At the time, the Korean Pharmaceutical Association strongly opposed the proposal, expressing concern that the unmanned sale of medications could expand gradually following the approval of video-based dispensing machines. As discussions failed to reach a conclusion for an extended period, the matter remained effectively on hold.However, during this latest meeting, the Ministry of Trade, Industry, and Energy reportedly emphasized that “a conclusion needs to be reached on the issue that remained unresolved for 5 years,” strongly advocating for the necessity of a pilot exemption.It is reported that the company presented an additional plan to focus installations on medically underserved areas, such as remote islands and mountainous regions, where access to pharmacies and convenience stores is limited, in addition to its existing proposal.It is also reported that they emphasized the stability of the technology, noting that biometric authentication and control systems had been further refined during the period when discussions were suspended.On the other hand, the Korean Pharmaceutical Association and the Ministry of Health and Welfare reaffirmed their opposition. They reiterated the existing logic that pharmaceutical sales should be premised on face-to-face medication guidance and safety management systems, and that expanded unmanned sales could lead to misuse and safety concerns.During the meeting, a heated debate reportedly continued over whether to proceed with the pilot exemption.An official who attended the meeting stated, “Toward the end of the meeting, the atmosphere even reached the point where we were about to decide on proceeding with the exemption via a show of hands, but the debate between supporters and opponents continued for a long time. Ultimately, the Ministry of Trade, Industry and Energy decided to refer the matter to the next expert committee.”At the meeting, ministry officials reportedly stated that a sixth expert committee meeting would be convened within 1-3 months to make a final decision on whether to proceed with the pilot exemption.Within and outside the industry, some observers believe a partially positive sentiment toward the project is forming inside the Ministry of Trade, Industry, and Energy. In fact, several participants at the meeting reportedly expressed opinions sympathetic to the need for pursuing the pilot project.However, given the continued strong opposition from the Ministry of Health and Welfare and pharmacy organizations, as well as the possibility that allowing unmanned pharmaceutical sales could expand into a broader social controversy, significant conflict is expected before a final conclusion is reached.

- Opinion
- ‘Need to change perceptions on menopausal hormone therapy’
- by Son, Hyung Min May 18, 2026 09:11am
- With the US Food and Drug Administration (FDA) recently removing the Black Box Warning imposed on menopausal hormone therapy (MHT) products, the likelihood of a shift in perception is growing within Korea’s obstetrics and gynecology clinical field.While there has long been a strong tendency to avoid treatment due to concerns about the risk of breast cancer and cardiovascular disease, there is now a growing emphasis on the need for personalized treatment that takes into account the patient’s age, time of menopause, and risk factors.In particular, experts pointed out that the findings of the US Women’s Health Initiative (WHI) study had been excessively generalized to women in early menopause, and stressed that the long-term health benefits of early treatment, such as the prevention of cardiovascular disease, osteoporosis, and dementia, should also be considered.In a recent meeting with DailyPharm, Eun Sil Lee, Professor of Obstetrics and Gynecology at Soonchunhyang University Seoul Hospital, and Tae-Hee Kim, Professor of Obstetrics and Gynecology at Soonchunhyang University Hospital Bucheon, assessed that this FDA action could serve as an opportunity to fundamentally redefine existing perceptions of MHT, going beyond the mere removal of warning labels.(From the left) Professor Eun Sil Lee (Obstetrics and Gynecology, Soonchunhyang University Seoul Hospital) and Professor Tae-Hee Kim (Obstetrics and Gynecology, Soonchunhyang University Hospital Bucheon)The two professors particularly pointed out that although the 2002 WHI study was conducted on a patient population different from actual women in early menopause, the findings were subsequently applied uniformly to women of all ages, creating excessive fear toward hormone therapy.Based on the study results, the FDA introduced Black Box Warnings for MHT products in 2003. Following this, concerns over breast cancer, cardiovascular disease, and dementia risks spread rapidly, causing hormone therapy prescriptions to decline sharply worldwide. However, as more age-specific and menopause timing-based reanalyses accumulated in recent years, the FDA initiated procedures last November to remove the warning.Experts point out that the WHI study results were overgeneralized.The WHI study included women with an average age of 63, many of whom already had a significant number of risk factors for cardiovascular disease. Another limitation cited is that the study used a combination of hormones that is no longer widely used today. Subsequent age-specific analyses showed that for women who began treatment within 10 years of menopause, particularly those in their 50s, there was no clear increase in the risk of cardiovascular disease or dementia, and some studies even suggested possible preventive effects.In practice, MHT has long been used as a representative menopause management therapy, having been proven effective not only in alleviating menopausal symptoms such as hot flashes, sleep disturbances, and depression but also in preventing osteoporosis. However, following the publication of the WHI study, concerns about breast cancer risk spread rapidly, leading to a strong trend in Korea toward discontinuing or avoiding the treatment.Recently, there has been growing discussion that MHT should be reevaluated from a “well-aging” perspective, one that goes beyond simple symptom management to include cardiovascular health, osteoporosis prevention, and the management of healthy life expectancy. As life expectancy increases and women spend longer periods living after menopause, experts argue that both risks and benefits of treatment should be evaluated in balance.The two professors emphasized, “MHT should not be oversimplified solely in terms of breast cancer risk. It should instead be approached from the perspective of personalized treatment that comprehensively considers patient age, menopausal timing, and overall health status. When treatment begins at the appropriate time, positive effects can be expected not only for quality of life improvement but also for long-term health management.”Q. How do you evaluate the FDA’s recent removal of the MHT Black Box Warning?Professor Tae-Hee Kim (Obstetrics and Gynecology, Soonchunhyang University Hospital Bucheon)Professor Tae-Hee Kim: I view this FDA action as an important opportunity to reevaluate the previously excessive perception of MHT risks based on evidence.The patients included in the WHI study had a median age of 63 and included women with cardiovascular risk factors. They differed from actual women in early menopause who typically begin hormone therapy. Moreover, the study used hormone combinations that are rarely used today, making it difficult to generalize the results to all patients.I think this decision carries significance in reorganizing overly emphasized risks so that women in early menopause who may benefit from hormone therapy do not avoid treatment excessively.”Professor Eun Sil Lee: The previous black box warning stated that prescribing MHT increased the risk of breast cancer, cardiovascular disease, and dementia, which significantly heightened patients’ fears. In fact, the frequency of hormone therapy use decreased significantly following the WHI study.However, subsequent age-specific analyses yielded different results. For women in their 50s who began treatment within 10 years of menopause, there was no clear increase in risk. On the contrary, the possibility of preventing cardiovascular disease or dementia was raised. Ultimately, this means that the timing of when hormone therapy is started is what matters.Q. How should the safety of MHT be evaluated?Professor Tae-Hee Kim: Previously, there was a strong perception that taking hormones increased the risk of cardiovascular disease and dementia, but in fact, these findings should be viewed as results from women who started treatment in their 60s and 70s.On the other hand, data is accumulating showing that starting treatment in one’s 50s, during the early stages of menopause, may actually have preventive effects against cardiovascular disease and dementia. Ultimately, the key issue is not simply whether or not to take hormones.Factors such as age, menopausal timing, and cardiovascular risk factors must all be considered comprehensively. I believe safety is determined by individualized treatment strategies tailored to each patient.Professor Eun Sil Lee: I believe the most important factors when evaluating safety are the patient’s age and the timing of menopause.In reality, women in the early stages of menopause often do not have a relatively high risk of cardiovascular disease. Rather, during this period, as hormone levels drop sharply, vascular health deteriorates, and changes such as decreased bone density, sleep disorders, and feelings of depression begin to manifest in earnest.Conversely, if a patient has already progressed into her 60s with advanced atherosclerosis, the approach may differ. In advanced atherosclerosis, hormone therapy could potentially affect thrombosis risk. In the end, the key issue is who starts treatment and when.In actual clinical practice, many patients with severe menopausal symptoms hesitated to pursue treatment due to fears such as ‘Won’t this increase my dementia risk?’ or ‘Won’t this cause cardiovascular disease?’ But recently, the concept of individualized treatment considering age, risk level, and menopausal timing has become increasingly important.Ultimately, I believe the safety of MHT is not an issue that can be explained in a one-size-fits-all manner; it must be assessed by comprehensively considering the patient’s health status and the timing of treatment.Q. How are differences in safety between products distinguished in actual clinical practice?Professor Eun Sil Lee: The approach to hormone therapy fundamentally differs depending on whether the patient has a uterus. Women without a uterus can use estrogen-only therapy, but women with a uterus must use progesterone in combination to prevent endometrial cancer.The characteristics of treatment differ depending on which progesterone is used. Appropriate therapy inevitably varies according to patient age, symptoms, risk level, and preference.Professor Tae-Hee Kim: Rather than saying a specific product is absolutely better, it is more accurate to view each hormone therapy as having distinct characteristics.It is important to select the most suitable medication by considering the patient’s lifestyle, symptoms, and health status. Ultimately, individualized treatment through consultation with a specialist is key.Q. How do you evaluate the breast cancer risk associated with MHT prescriptions?Professor Eun Sil Lee (Obstetrics and Gynecology, Soonchunhyang University Seoul Hospital)Professor Eun Sil Lee: In reality, differences exist depending on the medication. European studies showed that combinations of estrogen and natural progesterone did not demonstrate a clear increase in breast cancer, while some synthetic progesterone combinations showed tendencies toward increased risk.However, even if long-term use carries some increased risk, the absolute risk itself is interpreted as not very large. Above all, regular screenings are crucial.Women receiving hormone therapy tend to undergo regular screenings more consistently, and management is possible through early detection. Ultimately, I think accurate explanations are needed so patients do not abandon treatment based solely on vague fear.Professor Tae-Hee Kim: Many women have a vague fear that taking hormone therapy will cause breast cancer. But actual data show that the issue is not that simple.Even in the WHI study, women without a uterus did not show an increase, but rather a tendency toward reduced breast cancer incidence. European studies also found differences in breast cancer risk according to progesterone type. Some medications showed no significant increase.Of course, it cannot be said that taking hormones absolutely prevents breast cancer. However, the important point is that breast cancer mortality did not increase. In fact, overall mortality was lower.Benefits such as improved quality of life, fracture prevention, and cardiovascular disease prevention must also be considered together. Individualized treatment based on family history and risk level is important.Q. How do you predict MHT prescriptions will change in the Korean market going forward?Professor Tae-Hee Kim: I believe changes in perception toward MHT will clearly emerge in Korea as well. In particular, as life expectancy increases, interest is continuing to grow not only in living longer, but in aging healthily—that is, in ‘well-aging’ and ‘anti-aging.’Women now live for more than 30 to 40 years after menopause. Ultimately, how healthily this period is managed has become extremely important. From that perspective, hormone therapy should be viewed not merely as symptom control for hot flashes or sleep disorders, but as part of a healthy lifespan management strategy.Most important is the timing of treatment initiation. Starting treatment within 10 years after menopause or before age 60 is absolutely advantageous. Diseases such as cardiovascular disease, dementia, and osteoporosis become difficult to reverse once they progress. Therefore, we must approach them from a preventive perspective, which requires starting management from the early stages of menopause.In actual clinical practice, many patients experience major declines in quality of life due to osteoporotic fractures, fall risk, sleep problems, and joint pain. Hormone therapy can help improve these issues as well.Professor Eun Sil Lee: I believe the current prescribing environment is likely to expand further. However, rather than simply increasing prescriptions across the board, “personalized treatment” tailored to each patient’s characteristics will become more important.After menopause, vascular health deteriorates rapidly due to a decrease in estrogen. Atherosclerosis begins to progress, and bone density also decreases rapidly. Therefore, in women in the early stages of menopause, hormone therapy can play a positive role in preventing osteoporosis and maintaining vascular health.Conversely, the approach may differ for older women in whom atherosclerosis has already progressed significantly. Ultimately, this means that the patient’s age, vascular condition, and the timing of menopause must all be taken into account.Recently, the FDA has also emphasized the need for an approach that takes age and the timing of menopause into account. In fact, the FDA has recommended starting treatment within 10 years of menopause or before the age of 60.Most importantly, patient perception itself must change. Until now, the perception that ‘hormone therapy is always dangerous’ has been too strong. But now patients are beginning to think about both quality of life and healthy lifespan.Going forward, rather than simply enduring menopausal symptoms, interest in how to maintain health after menopause is likely to increase further. In that process, it will become important for patients to consult sufficiently with medical professionals and choose treatments suited to themselves.

- Company
- Curocell begins commercialization process for Korea's first CAR-T
- by Cha, Ji-Hyun May 15, 2026 02:44pm
- Curocell is pursuing market entry following the approval of South Korea's first domestically developed chimeric antigen receptor T-cell (CAR-T) therapy. Based on its domestic R&D and manufacturing infrastructure, the company aims to improve patient access to the CAR-T treatment landscape that has previously relied mostly on overseas facilities. Furthermore, the company plans to strengthen its next-generation pipeline through a strategy of expanding indications.On May 14, Curocell held a press conference at the Four Seasons Hotel in Seoul to commemorate the approval of its CAR-T therapy, 'Rimqarto Inj' (ingredient name: anbalcabtagene autoleucel). The company unveiled Rimqarto's clinical value, domestic commercialization strategy, and directions for subsequent clinical development. The event was attended by Curocell CEO Gunsoo Kim, Professor Won Seog Kim of Hemato-oncology at Samsung Medical Center, Curocell Executive Director Seungwon Lee, and Su-hee Cho, Head of Curocell’s Clinical Development Center.Rimqarto is an autologous CD19-targeting CAR-T therapy that uses a patient's own T cells. The process involves collecting T-cells from the patient's blood, introducing genes that allow them to recognize cancer cells, proliferating them ex vivo, and reintroducing them into the patient. It has been designed with Curocell's proprietary OVIS platform, designed to maximize therapeutic effects by simultaneously suppressing the expression of PD-1 and TIGIT, immune checkpoint receptors that inhibit the anticancer activity of T cells.Previously, on April 29, the Ministry of Food and Drug Safety (MFDS) granted marketing authorization for Rimqarto as a treatment for adult patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) and primary mediastinal large B-cell lymphoma (PMBCL) after two or more lines of systemic therapy. With this, Rimqarto has been listed as the 42nd novel drug developed in Korea and the first CAR-T therapy developed by a Korean company.Curocell CEO Gunsoo KimCEO Kim shared in his opening remark that "The approval of Rimqarto is more than just one new drug entering the market. Curocell has step-by-step built a foundation that allows the entire process(from R&D to clinical trials, production, quality control, and licensing) to be performed domestically."CEO Kim emphasized that Rimqarto is a treatment option that can expand patient access in the CAR-T treatment landscape, which has been highly dependent on overseas supply. "For patients with relapsed or refractory DLBCL, treatment options are limited and disease progression can be rapid, making timely treatment critical," and added, "While existing CAR-T therapies faced barriers before reaching Korean patients in clinical settings, we will do our best to improve treatment accessibility and establish a stable supply base through Rimqarto."Professor Won Seog Kim, the first presenter, explained the unmet needs in DLBCL treatment and the clinical value of Rimqarto. According to Professor Kim, approximately 6,000-6,500 cases of malignant lymphoma occur annually in Korea, with DLBCL being the representative disease, accounting for 40% of cases. However, 35–40% of DLBCL patients experience recurrence after standard treatment, and patients reaching the third-line treatment stage number around 700 annually in Korea, carrying a poor prognosis.CAR-T therapies are considered an option that has changed the treatment paradigm in this relapsed/refractory DLBCL field. Professor Kim said, "In a patient group where the possibility of long-term survival was previously around 10%, data showed that CAR-T treatment could offer another treatment opportunity. For this reason, we could proceed quickly to obtain reimbursement, and added, "However, even with current commercial CAR-T therapies, the expectation for a complete cure remains at about 40%, leaving room for further improvement."Professor Kim presented Curocell's proprietary OVIS technology as Rimqarto’s distinguishment. One reason existing CAR-T therapies fail is that T-cells become exhausted or immune checkpoint mechanisms such as PD-1 or TIGIT become active, preventing the sufficient elimination of cancer cells."Curocell’s OVIS technology works by loading small RNA fragments into the CAR-T to suppress the expression of immune checkpoints like PD-1 and TIGIT," Professor Kim explained, "Rimqarto is a differentiated, next-generation CAR-T product in that it can overcome treatment failure caused byover-expression of immune checkpoints."On May 14, Curocell held a press conference to commemorate the approval of its CAR-T therapy, 'Rimqarto Inj' Clinical data also confirms Rimqarto’s competitiveness. In Phase 2 trial, Rimqarto demonstrated an objective response rate (ORR) of 75% and a complete response (CR) rate of 67% based on independent review committee evaluations. Regarding safety, the incidence of Grade 3 or higher cytokine release syndrome (CRS) was 9%, and neurotoxicity was 4%. Professor Kim said, "The response rate of over 75% and a CR rate of 67% as evaluated by the committee are highly encouraging results. It showed data that is manageable not only in terms of efficacy but also safety."Starting with this approval, Curocell plans to accelerate the commercialization of Rimqarto. Related to the core pillars of the commercialization strategy, Executive Director Lee Seung-won presented ▲rapid insurance reimbursement listing ▲expanding patient accessibility. "As CAR-T therapies are perceived as high-priced treatments, the speed of reimbursement listing is directly linked to patient access," Lee explained, " Rimqarto was selected for the parallel 'Approval-Evaluation-Negotiation' pilot project, listed on a track that can shorten the time from approval to reimbursement by approximately 90 days." Curocell is currently responding to supplementary data requests for the drug reimbursement evaluation and designing price-negotiation scenarios to support a reimbursed launch as early as September.The company will also pursue its domestic production base to build a supply chain and expand patient access. Lee stated, "Existing global CAR-T therapies involve a structure where a Korean patient’s cells are sent to an overseas manufacturing site and then brought back, which takes weeks and carries international transport risks. Rimqarto can achieve a fast 'vein-to-vein' time based on its domestic production facility." Curocell plans to establish a system within the year allowing Rimqarto treatment at 30 hospitals nationwide and will manage the entire process (from ordering to collection, manufacturing, delivery, and administration) through its online tracking platform, 'CuroLink.'On May 14, Curocell held a press conference to commemorate the approval of its CAR-T therapy, 'Rimqarto Inj' The company is also strengthening its next-generation pipeline by expanding Rimqarto's indications. As a follow-up development strategy, Curocell is considering expanding indications to adult acute lymphoblastic leukemia (ALL), systemic lupus erythematosus (SLE), and second-line treatment for DLBCL. Director Cho Su-hee explained, "There are currently no clear treatment options after standard therapy for patients over the age of 25, who make up the majority of adult ALL patients," and added, "Curocell has been preparing to expand the indication to adult ALL since 2022 and is currently in the final stages of Phase 1." The company plans to enter Phase 2 shortly and is also pursuing an expansion of clinical trials in Japan to strengthen global capabilities.The company is also pursuing expansion into autoimmune diseases and earlier lines of treatment. Director Cho stated, "To provide a better life for patients with lupus nephritis, we have started Korea's first autoimmune disease CAR-T clinical trial. Based on the experience of administering Rimqarto to lupus nephritis patients through "approval for use for therapeutic purposes," we expect encouraging results," and added, "Since Rimqarto showed a high response rate compared to other agents in third-line DLBCL, moving it up to second-line treatment would help even more patients."