- LOGIN
- MemberShip
- 2026-07-27 14:04:03
- Policy
- 515 days required from GIFT designation to approval
- by Lee, Tak-Sun Jul 27, 2026 08:43am
- New drugs that received marketing authorization this year (2026) through the Ministry of Food and Drug Safety's 'Global Innovative products on Fast Track (GIFT)' program took an average of 515 days from the designation date to final approval. Although the MFDS has significantly shortened its actual review time, a significant amount of time was taken by the pharmaceutical companies to prepare and supplement extensive global approval data.Dailypharm’s analysis of 10 products (GIFT Nos. 50–59) that obtained marketing approval through the MFDS' GIFT program in 2026 found that the average elapsed time from GIFT designation to final approval was 515.5 calendar days.The fastest approval was achieved by ‘Wainua Autoinjector (eplontersen),’ developed by AstraZeneca Korea for transthyretin-mediated amyloidosis. The product received marketing approval in July 2026, just 305 days after receiving GIFT designation in September 2025.In contrast, Verto Korea’s orphan drug ‘Joenja Tab’ recorded the longest timeline at 801 days, from its GIFT designation in April 2024 to final approval. Other products that took more than a year and a half included ‘Lamzede Inj’ from Kwangdong Pharm (767 days) and ‘Rimqarto Inj’ from Curocell (631 days)l, Korea's domestically developed CAR-T therapy.Time from GIFT designation to approval for key products approved in 2026The average timeline of approximately 17 months (515 days) is largely affected by the period required for companies to prepare and submit supplemental data. In other words, the entire period between GIFT designation and final approval should not be interpreted as a delay in the MFDS review process. Nevertheless, further efforts to minimize the need for additional submissions will be necessary to ensure that patients gain faster access to innovative medicines.Through the GIFT program, the MFDS aimed to shorten the statutory review period by 25%, completing reviews within 90 working days instead of the standard 120 working days. In practice, the agency's actual review time has been shortened, averaging around 60 to 70 days.The challenge arises when the MFDS requests additional information during the review process, at which point the statutory review clock stops. Industry sources noted that because many GIFT-designated products are innovative medicines from global pharmaceutical companies or advanced biopharmaceuticals, it often takes several months, or even more than a year, to coordinate GMP inspections at overseas manufacturing sites, conduct additional analyses of multinational clinical data, complete quality verification, and prepare supplementary materials in consultation with global headquarters.Building on the expertise gained through the GIFT program's ‘expedited review and rolling review’ system, the MFDS is also accelerating efforts to shorten approval timelines for all new drugs.After reducing the average review period for new drug approvals from approximately 420 days to 295 days, the agency introduced a new 240-day review framework in 2026, which is one of the fastest in the world. To support this initiative, it significantly expanded its review workforce and replaced the previous sequential review structure for nonclinical, clinical, and quality assessments with a parallel review system conducted simultaneously across departments.The MFDS has also institutionalized at least two ‘pre-submission face-to-face meetings ‘ before companies file marketing authorization applications to reduce approval delays caused by requests for additional information. By providing guidance and checklists in advance to improve documentation quality, the agency aims to minimize clock-stops during the review process.An MFDS official said, "We are fundamentally transforming the drug review system to create an environment in which patients can gain access to new medicines faster than anywhere else in the world. We will continue strengthening the predictability and transparency of the GIFT program while working closely with industry so that companies can bring innovative medicines to market without the burden of extensive supplementary submissions."

- Company
- Calls for reform in NHI access for third-line mCRC treatment
- by Son, Hyung Min Jul 27, 2026 08:43am
- There is a growing demand in South Korea to improve access to third-line treatments under the National Health Insurance (NHI) for metastatic colorectal cancer (mCRC) to enhance patient survival.While health insurance coverage is relatively comprehensive for first- and second-line treatments in South Korea, global standard-of-care agents used in subsequent lines remain non-reimbursed. In response, health authorities have acknowledged the high unmet medical need in third-line treatment, stating that they are evaluating measures to improve patient access.On the 24th, a symposium titled "Policy Forum for Improving the Treatment Environment for Metastatic Colorectal Cancer Where Early Treatment Access Determines Survival" was held at the National Assembly, hosted by Representative Mihwa Seo of the Democratic Party of Korea.Rep. Seo stated, "In metastatic colorectal cancer, treatment timing and therapeutic access exert a direct impact on overall survival, yet reimbursed options in third-line are not available in South Korea," and added, "A rational reimbursement framework must be established to prevent patients from forfeiting treatment due to financial toxicity."A policy symposium on improving the treatment environment for metastatic colorectal cancer was held on July 24 at the National Assembly Members' Office Building.Third-line treatments are not reimbursed...Leading to drops in treatment ratesPresenting at the forum, Professor Myung Ah Lee of the Division of Oncology at Seoul St. Mary's Hospital diagnosed that as patients with metastatic colorectal cancer progress through sequential lines of therapy, acquired resistance and disease progression progressively deteriorate their systemic performance status. Dr. Lee explained that non-reimbursed drug costs starting at the third-line setting lead to a sharp decline in the proportion of patients maintaining ongoing treatment.Professor Lee pointed out, "Reimbursement is well-integrated through first- and second-line systemic chemotherapy, resulting in a manageable financial burden for patients. However, from the third-line setting onward, patients with good performance status are frequently unable to receive treatment simply because no reimbursed agents exist," and added, "An increasing number of patients are discontinuing treatment despite viable therapeutic opportunities due strictly to financial constraints."Currently, first-line therapy for metastatic colorectal cancer primarily utilizes oxaliplatin- or irinotecan-based cytotoxic chemotherapy in combination with targeted biologics. Second-line treatment involves switching to the alternative chemotherapy backbone not administered in the first-line setting.However, the treatment landscape shifts dramatically following failure of both lines. While a small subset of patients with specific biomarkers can access immune checkpoint inhibitors or targeted agents, the eligible patient population remains narrow.For the majority of patients, global standard-of-care regimens recommended in guidelines, such as the combination of 'Lonsurf (trifluridine/tipiracil)' plus 'Avastin (bevacizumab)', 'Stivarga (regorafenib)', and 'Fruzaqla (fruquintinib)', are all non-reimbursed in South Korea.Professor Lee emphasized, "In the U.S. and Europe, Stivarga, Lonsurf-Avastin combination, and Fruzaqla are recommended from the third-line setting, but none of these regimens are reimbursed in South Korea," and added, "We need a regulatory environment where therapeutics capable of preserving quality of life alongside overall survival can enter the national health insurance benefit umbrella more rapidly."Access to Next-Generation Sequencing (NGS) testing for precision oncology was also pointed out to be resolved, as the absence of genomic profiling data restricts patient eligibility for biomarker-driven novel drugs and clinical trial enrollment.Professor Lee stated, "While international practice is moving toward routine genomic profiling for patients with metastatic or recurrent solid tumors, domestic reimbursement in Korea remains restricted outside of specific cancer types," and expressed concerns that "Consequently, patients who wish to participate in clinical trials for novel therapies often cannot enroll due to a lack of genetic sequencing results."Experts urged that even in third-line and subsequent settings, clinicians must have the therapeutic flexibility to decide the sequence of care based on patient performance status, prior treatment history, and specific toxicity profiles.Professor Dong-Hoe Koo of the Division of Hematology-Oncology at Kangbuk Samsung Hospital explained, "Each therapeutic option possesses distinct efficacy as well as unique toxicity profiles, such as fatigue, thrombocytopenia, hand-foot syndrome, and hypertension,” and added, “Drug selection should be personalized according to prior treatment exposure and individual patient vulnerability to specific adverse events."Dr. Koo added, "Because third-line therapeutics established as global standards remain non-reimbursed in Korea, a patient's financial status directly dictates therapeutic choices," and added, "Reimbursement access must be improved so that patients with preserved performance status can gain survival opportunities through third-line and subsequent therapies."(From left) Min-Jung Kim, Administrative officer at the Ministry of Health and Welfare; So-Young Lee, Manager of the Pharmaceutical Benefit Management Division at HIRA; Professor Dong-Hoe Koo of Kangbuk Samsung Hospital; and Professor Myung Ah Lee of Seoul St. Mary's Hospital.Discussions continue on regulatory reforms to address unmet medical needDuring the panel discussion, structural limitations in the pharmacoeconomic evaluation process and directions for regulatory reform emerged as key agenda items.Reporter Yun-Ho Eo of DailyPharm pointed out that novel therapeutics approved via placebo-controlled clinical trials face structural disadvantages during pharmacoeconomic evaluations, as they are forced to compete against outdated comparator drugs.Eo stated, "Recently, we have observed significant delays between passing the Cancer Disease Review Committee (CDRC) and being tabled before the Pharmaceutical Reimbursement Evaluation Committee (PREC)," and added, "Special policy mechanisms need to be considered for diseases where a new treatment landscape has formed, but cost-effectiveness is inherently difficult to prove due to outdated comparator drugs."Eo added, "Even if a drug does not qualify for a full pharmacoeconomic evaluation waiver or an elevated Incremental Cost-Effectiveness Ratio (ICER) threshold, we need flexible regulatory pathways for drugs occupying an intermediate tier," and added, "Multinational pharmaceutical subsidiaries in Korea must also actively negotiate with their global headquarters rather than abandoning reimbursement due to challenging external environments."Government representatives acknowledged the severity of the coverage gap in third-line metastatic colorectal cancer. They explained that regulatory reforms are underway to incorporate high unmet medical needs into reimbursement decision-making.Lee stated, "We are aware of the reality that patients face due to a lack of options in third-line therapy, and we feel a deep sense of responsibility," and added, "Under the principle that unmet medical needs in life-threatening severe diseases should be evaluated through dedicated mechanisms, we are accelerating regulatory reforms."The government currently operates a "conditional early listing with post-evaluation" pathway for high-cost novel drugs that demonstrate substantial unmet medical need despite limited clinical evidence. Separately, health authorities are evaluating flexible ICER thresholds for therapies indicated for severe diseases where applying standard ICER benchmarks is unfeasible.Lee stated, "A research initiative evaluating the application of flexible ICER thresholds is scheduled for completion around November of this year, after which implementation will proceed," and added, "Even before the completion of this research, working-level staff is thoroughly reviewing data so that the evaluation committee can adequately consider disease characteristics and unmet needs."Lee noted, "In reviewing third-line treatments, including Fruzaqla, we are re-examining the reasons why previous agents failed to secure reimbursement, the criteria applied at the time, and our newly evolved administrative procedures," and added, "Even before the research findings are published, we will fully consider the third-line colorectal cancer reimbursement gap within the committee's existing criteria and authority."Min-Jung Kim, Manager of the Division of Health Insurance Benefits at the Ministry of Health and Welfare, noted, "Because National Health Insurance operates within a finite budget, we must strike a balance between patient access and fiscal sustainability," and added, "We are striving to establish rational reimbursement solutions by comprehensively reviewing the clinical value of therapeutics and their impact on patient quality of life."Kim added, "It is important for pharmaceutical companies to demonstrate proactive negotiation and a willingness to improve patient access," and concluded, "The government will continue driving regulatory improvements to ensure that essential therapeutics are supplied to patients more rapidly."

- Policy
- Differentiated R&D investment ratio applied to innovative pharma
- by Lee, Jeong-Hwan Jul 27, 2026 08:43am
- AI-generated imageThe Ministry of Health and Welfare (MOHW) is implementing the amendment to the "Enforcement Decree of the Special Act on Designation and Support of Pharmaceutical Industry," establishing differentiated annual research and development (R&D) investment thresholds for "Innovative Pharmaceutical Company" certification based on corporate revenue scale and global manufacturing capabilities.Under the revision, variable R&D investment ratios ranging from 7% to 9% will be applied using an annual revenue benchmark of KRW 100 billion. Notably, companies that have secured advanced Good Manufacturing Practice (GMP) certifications from the United States or Europe will be eligible for a lowered threshold of 5%, which is expected to ease the certification burden for drugmakers expanding into international markets. On July 24, according to biopharmaceutical industry sources, the amended Enforcement Decree, which took effect on the 21st, codifies the specific R&D expenditure requirements into three distinct categories under Article 2-2 ("Annual Research and Development Expenditure Requirements"). Differentiated 7–9% R&D sizes based on KRW 100 billion criterion…5% threshold for global GMP holdersFirst, small and medium-sized enterprises (SMEs) and mid-tier pharmaceutical companies with annual pharmaceutical revenues under KRW 100 billion must invest either at least KRW 7 billion annually or at least 9% of their annual pharmaceutical sales in R&D to meet the certification criteria.In contrast, large-scale pharmaceutical enterprises with annual pharmaceutical revenues of KRW 100 billion or more must allocate at least 7% of their annual sales to R&D to qualify for the Innovative Pharmaceutical Company designation. A notable feature of the amendment lies in the relaxed benchmark for companies equipped with global manufacturing capabilities. Pharmaceutical enterprises holding Good Manufacturing Practice approvals (such as US cGMP or EU-GMP) from regulatory authorities in the United States (FDA) or the European Union (EMA) will see their required R&D investment threshold lowered to 5% of annual pharmaceutical revenue. This provision effectively recognizes extensive investments in world-class manufacturing infrastructure as an innovative activity equivalent to direct R&D. Averaged sales from three previous fiscal year…emphasizing accounting transparencyThe amendment also clarifies the specific accounting principles and calculation methods for R&D expenditures and sales. Only revenues and expenses directly tied to "pharmaceutical products" as defined under the Pharmaceutical Affairs Act will be recognized, explicitly excluding performance metrics from non-pharmaceutical business divisions such as health functional foods or cosmetics.To prevent administrative confusion arising from short-term financial volatility, compliance will be evaluated based on the average pharmaceutical R&D expenditure and average pharmaceutical sales over the "immediately preceding three fiscal years," counting back from the fiscal year in which the company submits its certification application. For newly established companies operating for less than three years, metrics will be annualized based on performance generated up to the date of application.To enhance financial transparency, the amendment mandates that accounting for qualifying R&D expenses must strictly adhere to statutory accounting standards established under the Act on External Audit of Stock Companies. Details regarding specific expense items eligible for inclusion under pharmaceutical R&D costs will be outlined in a separate administrative notification issued by the Minister of Health and Welfare.Following the enforcement of this decree, the MOHW is expected to finalize the broader structural amendment of the Innovative Pharmaceutical Company certification system.
- Company
- Voxzogo may be prescribed at tertiary hospitals in Korea
- by Eo, Yun-Ho Jul 27, 2026 08:43am
- Voxzogo, Korea’s first treatment for children with achondroplasia, has gained a foothold in general hospitals in Korea.According to industry sources, Voxzogo (Vosoritide), which Samoh Pharm licensed from BioMarin Pharmaceutical, has been approved by the drug committees (DCs) at 10 hospitals nationwide, including four of Korea's ‘Big 5’ hospitals—Samsung Medical Center, Seoul National University Hospital, Asan Medical Center, and Severance Hospital—as well as Keimyung University Dongsan Medical Center and Chungnam National University Hospital.Following its inclusion on the National Health Insurance reimbursement list last month, the therapy has been rapidly expanding its prescribing footprint.Voxzogo targets the FGFR3 signaling pathway, which is involved in the underlying cause of achondroplasia. In Korea, it was designated as the 10th product under the Ministry of Food and Drug Safety's Global Innovative Products on Fast Track (GIFT) program and approved for pediatric patients aged 4 months or older with achondroplasia with open growth plates.Under the reimbursement criteria that took effect in June, eligible patients are children aged 4 months or older with a confirmed FGFR3 mutation by genetic testing. Patients must have open growth plates and must not have undergone limb-lengthening surgery.To continue treatment, patients are evaluated every 6 months. Therapy is discontinued if the growth plates close or if the patient's annual growth velocity fails to meet the required threshold.Achondroplasia is not simply a condition characterized by short stature. In addition to impaired growth, patients may experience disproportionate body proportions, orthopedic complications, neurological complications, respiratory problems, limitations in daily functioning, and psychosocial burdens.Accordingly, the goal of treatment extends beyond increasing height. The therapy holds significance in that it improves growth while lowering the burden of long-term health management, thereby improving the quality of life for patients and caregivers.Meanwhile, Voxzogo demonstrated its efficacy in a Phase III clinical trial. In a study involving 121 pediatric patients with achondroplasia aged 5 to 14.9 years, the Voxzogo group showed an increase in annualized growth velocity of 1.40 cm/year from baseline after 52 weeks of treatment, whereas the placebo group experienced a decrease of 0.17 cm/year, demonstrating a statistically significant improvement of 1.57 cm/year.

- Opinion
- ‘GSK will continue bringing innovative drugs to Korea’
- by Son, Hyung Min Jul 27, 2026 08:43am
- This year marks the 40th anniversary of GSK Korea. Since its establishment in 1986 with a focus on infectious disease prevention and basic public health, the company has steadily reshaped its portfolio, expanding from respiratory diseases and immunology to infectious diseases and, more recently, oncology.Korea is one of the key countries where GSK concentrates its R&D capabilities. Last year, GSK Korea conducted a total of 60 research projects, including global clinical trials, post-marketing surveillance studies, non-interventional studies, and investigator-initiated trials. Approximately 5,838 Korean patients participated in these studies, while the company's R&D investment reached KRW 31.7 billion during the same period.Approaching both the first anniversary of his appointment and the company's 40th anniversary, Gunnar Riediger, who assumed leadership of GSK Korea in August last year, outlined a vision that positions Korea as more than simply a commercial market for its pharmaceuticals. Riediger said the company aims to expand opportunities for Korean healthcare professionals and patients to participate in global new drug development while strengthening its role in bringing innovative medicines to Korean clinical practice more quickly.At a recent meeting with reporters, General Manager Riediger said Korea is among the key countries executing GSK's global strategy, expressing his stance to expand clinical development and the introduction of innovative medicines continuously. In addition, he presented plans to continue investment in the Korean market, focusing on respiratory and immunological disorders, oncology, HIV, and vaccines."Korea has world-class healthcare capabilities"...Recognized as a key clinical development hubGunnar Riediger, General Manager of GSK KoreaRiediger said the most impressive aspect of Korea during his first year has been its world-class healthcare infrastructure and research capabilities.He believes these strengths are further enhancing Korea's role in the global new drug development process."Reflecting on my experience over the past year, Korea's healthcare infrastructure has been impressive. The country possesses world-class human resources including medical experts, healthcare professionals, and clinical trial researchers. Together, these strengths create an outstanding environment for carrying out global clinical development projects.”GSK is currently conducting around 70 clinical development programs in Korea, primarily in oncology and immunology. The portfolio spans both early- and late-stage development, and Korea is regarded as one of the company's top 10 countries in its global clinical development strategy.Riediger said. "Korea is an important strategic market for GSK from a clinical research perspective, Its capability to conduct research across the full spectrum of development, from early-stage to late-stage clinical trials, means it will continue to play a significant role in our global R&D efforts."He also emphasized that innovative medicines must ultimately reach patients, and that fostering the environment is as important as R&D.Riediger said, "Innovation should not end at R&D. What matters is ensuring that patients can actually benefit from it. Going forward, GSK Korea will continue working with the government, the medical community, and other stakeholders to improve patient access to innovative medicines and contribute to the advancement of Korea's healthcare."Expanding portfolio around core therapeutic areas..."We will continue introducing innovative medicines"Riediger said GSK Korea will continue introducing new medicines and expanding indications in line with GSK's global R&D strategy, focusing on the company's core therapeutic areas.GSK's key R&D focus areas include respiratory, immunology and inflammation, oncology, HIV, and infectious diseases and vaccines. According to Riediger, GSK Korea is likewise expanding its research activities and product portfolio in line with the company’s global strategy.Over the past year, the company has made notable progress in hematologic malignancies. Omjjara (momelotinib), a treatment for myelofibrosis, secured National Health Insurance reimbursement listing following its launch in Korea, while Blenrep (belantamab mafodotin), an antibody-drug conjugate (ADC) for multiple myeloma, was also introduced to the Korean market.Riediger said, "What we have achieved for Korean patients over the past 12 to 18 months demonstrates that GSK's strategy of focusing on core therapeutic areas is translating into tangible outcomes. While continuing lifecycle management through indication expansions for existing products, we are also preparing to introduce new treatment options in solid tumors and hepatitis B."He added, " As the approximately 70 clinical development projects currently underway begin to bear fruit, Korean patients will continue to gain access to new treatment options over the coming months and years."Going forward, GSK Korea is also expected to play an expanded role in disease prevention. The company has long established a strong presence in Korea's pediatric vaccine market through participation in the National Immunization Program (NIP). However, as Korea enters a super-aged society, the importance of vaccination for adults and older populations is increasing, while government support remains largely focused on children.These demographic changes present both new opportunities and new challenges for GSK Korea. The company has built an adult vaccination portfolio that includes the RSV vaccine ‘Arexvy’ and the shingles vaccine ‘Shingrix.’ Alongside maintaining its competitiveness in pediatric vaccines, expanding awareness of the clinical value and societal importance of adult immunization and fostering the market for vaccination among older adults will be key priorities under Riediger’s leadership.Riediger said the role of vaccination must evolve as Korea enters a super-aged society. He explained that the national vaccination policy should expand beyond children's immunization to address the emerging healthcare needs of an aging population."GSK has long contributed to Korea’s National Immunization Program. Based on the available data, we have continued to emphasize that policymakers need to pay greater attention to the evolving healthcare needs of an aging population. A prevention-focused approach must be reflected in national healthcare policies and strategies.""Vaccination is no longer limited to preventing infectious diseases. Its broader value, including reducing caregiver burden, lowering healthcare costs, decreasing hospitalizations, and improving quality of life, should also be taken into account. Particularly in a country like Korea, which has transitioned into a super-aged society, a prevention-focused approach can serve as an important foundation for the healthcare system."
- Company
- Invossa still mired in legal battles 7 years after license revocation
- by Kim, Jin-Gu Jul 24, 2026 08:21am
- Kolon TissueGene's knee osteoarthritis candidate TG-C failed to demonstrate improvements in pain and physical function in its U.S. Phase III trial. The clinical setback is expected to have a significant impact on the ongoing litigation involving Invossa Invossa-K Inj (Invossa), which is being pursued against Kolon TissueGene and Kolon Life Science. The two companies are currently involved in a total of 55 Invossa-related lawsuits with claims exceeding KRW 100 billion.Kolon TissueGene and Kolon Life Science in 55 damages lawsuits worth KRW 104.1 billionAccording to Korea's Financial Supervisory Service on July 23, Kolon TissueGene and Kolon Life Science are currently involved in 55 lawsuits related to Invossa—32 involving Kolon Life Science and 23 involving Kolon TissueGene. Most are claims seeking damages related to Invossa, with the total amount in dispute near KRW 104.1 billion.The plaintiffs who filed the lawsuit claim that they suffered physical and financial damages due to the change in Invossa's main cell line, which was revealed in 2019.Invossa drew global attention after receiving approval from Korea's Ministry of Food and Drug Safety (MFDS) in July 2017 as the world's first gene therapy for knee osteoarthritis. TG-C is the product's US development name.However, during the US Phase III trial in March 2019, it was discovered that one of the product's principal components, originally described as cartilage-derived cells, had in fact been replaced with kidney-derived cells that carry tumorigenic potential. The MFDS revoked Invossa's marketing authorization in April 2019, while the US Food and Drug Administration (FDA) placed the clinical trial on hold. The FDA later lifted the clinical hold in April 2020.Kolon TissueGene is currently facing 16 shareholder lawsuits seeking damages for investment losses. A total of 2,048 shareholders are participating in these cases, with claims amounting to KRW 55.5 billion. In addition, 931 patients who received Invossa have filed 6 damages lawsuits against Kolon TissueGene for KRW 12.2 billion.Kolon Life Science is also engaged in large-scale litigation with both shareholders and patients. Shareholders have filed 21 lawsuits seeking KRW 24.1 billion in damages, while 941 patients have filed 10 lawsuits seeking KRW 12.4 billion. The company is also being sued by multiple domestic insurers seeking reimbursement through subrogation claims.Industry and legal experts expect the latest US Phase III results to have little direct impact on the outcome of the damages lawsuits. However, they say the failed trial could increase the companies' financial and legal burdens when courts determine damages or during settlement negotiations.Most cases still await first-instance rulings at 7 years…Kolon loses first patient lawsuitAlthough more than 7 years have passed since the first wave of litigation began, the vast majority of cases have yet to receive even a first-instance ruling.A recent damages lawsuit filed by Invossa patients, however, resulted in the first trial court decision. On July 9, the Seoul Central District Court ruled entirely in favor of 139 patients who sued Kolon TissueGene and Kolon Life Science for damages.The court found that Invossa had been manufactured using kidney-derived cells rather than the cartilage-derived cells identified in the original marketing application, recognizing this as a “manufacturing defect.” It also concluded that marketing and selling the product while labeling it as containing cartilage-derived cells violated both the Pharmaceutical Affairs Act and the Act on Fair Labeling and Advertising. Accordingly, the court held the companies liable for both economic damages and emotional distress suffered by the patients. It rejected Kolon's argument that the defect could not have been identified based on the scientific knowledge available at the time of manufacture.Despite prevailing at trial, it will take more time for patients to receive compensation, as Kolon has appealed the ruling to the Seoul High Court. In addition, Kolon has blocked enforcement actions such as the seizure and collection of claims by patients by filing for a stay of execution.Most shareholder lawsuits likewise remain at the first instance stage. Three lawsuits against Kolon TissueGene have already concluded after plaintiffs withdrew their claims or courts recommended settlement. In 4 of the company's 16 shareholder lawsuits, however, trial courts ruled in favor of Kolon. The plaintiffs have appealed, with those cases now before the appellate court.By contrast, the dispute with Mitsubishi Tanabe Pharma over the return of upfront payments and damages arising from the terminated licensing agreement was resolved early on. In 2016, Kolon Life Science signed a technology licensing agreement with Mitsubishi Tanabe worth up to JPY 50 billion and received an upfront payment of JPY 2.5 billion. Mitsubishi Tanabe terminated the agreement in December 2017 and filed arbitration with the International Chamber of Commerce (ICC) the following April. After the changed cell line issue emerged in May 2019, it was added as an additional ground for termination. Ultimately, Kolon returned the JPY 2.5 billion upfront payment along with JPY 134 million in damages in April 2021.Administrative appeal over ‘Invossa license revocation’ still pending before Supreme CourtKolon Life Science is also pursuing administrative litigation in addition to the civil lawsuits. After the Ministry of Food and Drug Safety (MFDS) revoked Invossa's marketing authorization in 2019, the company filed an administrative lawsuit challenging the decision.Both the Seoul Administrative Court (first instance) in February 2021 and the Seoul High Court (second instance) in February 2024 ruled in favor of the MFDS. The courts found that the discrepancy between the cell component described in the marketing authorization application and the one actually detected constituted a material defect, dismissing Kolon's claims. Kolon subsequently appealed to the Supreme Court, where the case remains pending.In addition to the lawsuit over the revocation of Invossa's marketing authorization, Kolon Life Science also filed lawsuits seeking to ▲overturn the MFDS Commissioner's revocation of its clinical trial authorization, ▲ invalidate the Daejeon Regional Office of Food and Drug Safety's order to recall and dispose of Invossa, and ▲cancel the Ministry of Health and Welfare's and the Ministry of Science and ICT's orders to recover government research funding.Among these, the lawsuits challenging the revocation of the clinical trial authorization and the recall and disposal order were voluntarily withdrawn by Kolon Life Science. Meanwhile, the lawsuit seeking to overturn the government's recovery of research funding ultimately ended in Kolon's favor after reaching the Supreme Court. The company had received KRW 1.25 billion in government funding during the development of Invossa.Executives including former Chairman Woong-yeol Lee and CEO Woosok Lee were acquitted in criminal casesCriminal proceedings against Honorary Chairman Woong-yeol Lee, former CEO Lee Woo-seok, and other Kolon Life Science executives concluded with acquittals.Honorary Chairman Woong-yeol Lee and former CEO Lee Woo-seok were indicted on charges including violations of the Capital Markets Act and the Pharmaceutical Affairs Act. Prosecutors alleged that they concealed the US Food and Drug Administration's clinical hold order, attracted investment through the company's listing, manipulated the share price, and made false disclosures in violation of capital markets regulations.Both the first- and second-instance courts acquitted the defendants. The trial court found that the evidence presented was insufficient to conclude that the defendants knowingly concealed the cell line change or intentionally made false disclosures to attract investment.The appellate court (second instance) ruling was also handed down in February of this year. The Seoul High Court also upheld the acquittals. The appellate court ruled that the misunderstanding regarding the origin of the cells was recognized only after the product had already been manufactured and marketed, characterizing it as an error made during the development process rather than a deliberate cover-up. The acquittals became final after prosecutors decided not to appeal to the Supreme Court.The former head of Kolon Life Science's Bio New Drug Research Center and its former medical team leader were also acquitted by the Supreme Court. They had been charged with obstruction of official duties by fraudulent means, fraud under the Act on the Aggravated Punishment of Specific Economic Crimes, and violations of the Subsidies Management Act.The trial court ruled that there was insufficient evidence to conclude that they had interfered with the MFDS's review process or fraudulently obtained government R&D subsidies by deceiving government evaluators. However, the former medical team leader was found guilty of providing entertainment to an MFDS official and was fined KRW 10 million. The appellate court likewise found no evidence of intentional submission of false data or subsidy fraud, and the Supreme Court upheld those findings by dismissing the prosecution's appeal. The bribery conviction, however, was ultimately upheld, leaving the fine in place.
- Policy
- “Division of industry within the MOHW”…fostering pharma biotech
- by Lee, Jeong-Hwan Jul 24, 2026 08:21am
- Sung-il Oh, the Head of the Division of Pharmaceutical and Biotech Industry"I consider the Division of Pharmaceutical and Biotech Industry as the Division of Industry within the Ministry of Health and Welfare. While the traditional duties of the Office for Healthcare Policy on regulatory laws under the Medical Services Act and the Pharmaceutical Affairs Act, the primary mission of the Division of Pharmaceutical and Biotech Industry is fundamentally to promote and foster technology, markets, and the industry. My priority is to listen extensively and meet as frequently as possible with pharma companies, biotech firms, and healthcare sector stakeholders to build an environment where the industry can perform at its best."Core mission of the Division of Pharmaceutical and Biotech Industry under the Ministry of Health and Welfare (MOHW) include fostering domestic blockbuster novel drugs, providing policy support for the pharmaceutical and biotech sectors to address supply instabilities of essential medicines, modernizing the Innovative Pharmaceutical Company certification system, and nurturing the global pharmaceutical and biotech industry.Sung-il Oh, newly appointed as the Head of the Division of Pharmaceutical and Biotech Industry on July 22, clearly understood the aim of the division and its key responsibilities, despite returning to healthcare administration after a hiatus.Meeting with the press that day, Oh explained that while the current government, including the MOHW, considers strengthening public access to healthcare and enhancing patient convenience a major pillar of healthcare policy, it also recognizes the promotion and development of the pharmaceutical and biotech industry as a vital task to achieve.In outlining his aspirations as the new division head, Oh pledged to "listen as much as possible and meet as many people as possible." As the administration has defined the pharmaceutical and biotech industry as a key growth driver for the nation's future and signaled regulatory rationalization for its fostering and promotion, his commitment signifies an intention to directly observe and hear what regulatory improvements and policies the market actually requires.Oh highlighted, "The current government has a very keen interest in promoting the pharmaceutical and biotech industry, something I observed during my previous role as the Regulatory Reform and Legal Affairs Officer," and added, "Unlike the previous administration where the Regulatory Reform Committee functioned under the Prime Minister, the current administration's Regulatory Rationalization Committee is chaired directly by the President. Biotech has consistently been a major agenda item at presidential meetings."Oh continued, "The advice from senior officials was to listen first. I am personally experiencing that the pharmaceutical and biotech industry has many factors distinct from general healthcare administration, and I am studying various issues extensively." He stated, "I maintain an open-minded approach toward everyone who comes to visit. For now, my focus will be on properly executing well-crafted existing policies, such as the reform plan for the Innovative Pharmaceutical Company criteria."Oh concluded by stating, "Government interest in regulatory rationalization is high, and as the government believes that promoting the pharmaceutical and biotech industry will expand the nation's future growth engines, there seems to be deep deliberation at the top," and added, "While general healthcare administration primarily deals with occupational disputes and conflict mediation, and national health insurance focuses on fee adjustments, the Division of Pharmaceutical and Biotech Industry is a unit built to promote and foster. Perspective needs to shift significantly. I intend to learn thoroughly right from the basics."
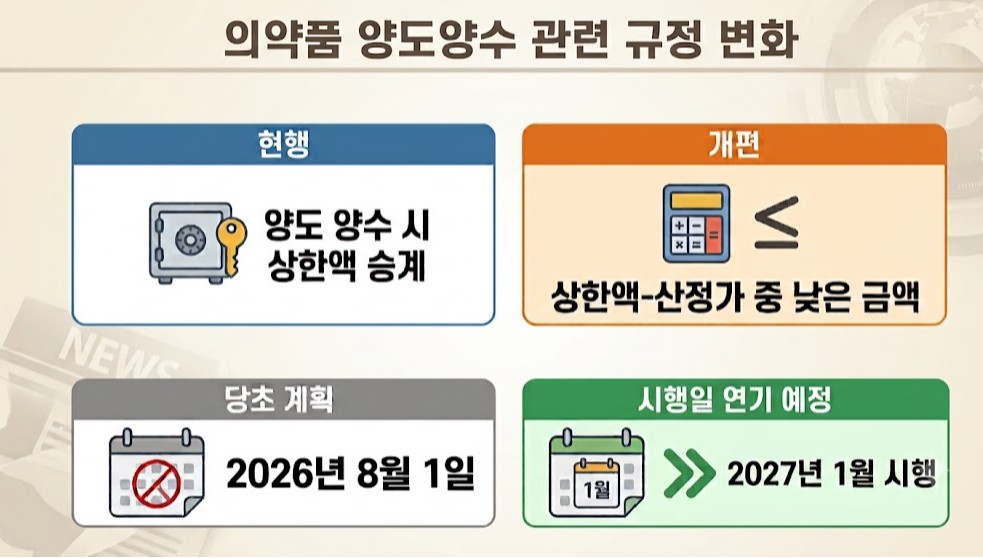
- Policy
- Ban on succession of price caps deferred until next year
- by Jung, Heung-Jun Jul 24, 2026 08:21am
- The government will postpone implementation of a new rule blocking the succession of reimbursement price caps during pharmaceutical product transfers and acquisitions until next year, following a grace period through the second half of this year.The change regarding price cap succession in product transfers was not included in the drug pricing reform agenda reviewed by the Health Insurance Policy Deliberation Committee (HIPDC) in March. The government has likely decided to delay implementation in consideration of on-site acceptability.Discussions on improving the implementation of the revised drug transfer and acquisition rules took place during a public-private consultative meeting involving the Ministry of Health and Welfare, the Health Insurance Review and Assessment Service (HIRA), the National Health Insurance Service (NHIS), and industry representatives on July 22.AI-generated imageThe revision to the drug product transfer and acquisition system was included in the Ministry’s May amendment to the 'Standards for Drug Pricing Decisions and Adjustments.' Until now, for products transferred between companies, the same reimbursement price ceiling as before the transfer was applied.Under the revised system, however, products acquired through transfers will instead receive whichever is lower—the newly calculated reimbursement price or the existing price ceiling—to prevent companies from using transfers to circumvent the revised drug pricing system.Because the generic drug pricing rate will change under the new pricing framework, transferred products will effectively be subject to the newly calculated reimbursement price rather than retaining the previous reimbursement ceiling.The rules governing pharmaceutical product transfers have undergone several revisions over the years. Since 2021, however, transferred products have generally been allowed to inherit the existing reimbursement price ceiling.Because the Ministry had initially planned to implement the revised notification on Aug. 1 following its administrative notice in May, the timeline was considered too tight, prompting agreement to introduce a grace period.With implementation now postponed until January next year, products currently under transfer negotiations are expected to proceed without disruption.A wave of last-minute product transfers is expected as companies reorganize their product portfolios in the second half of the year in response to the broader drug pricing reform,Since this will be the final grace period during which acquiring companies can still inherit the existing reimbursement price ceiling, transfer activity is expected to become more active than usual.

- Policy
- Post-listing price cuts to be made every April and October
- by Jung, Heung-Jun Jul 24, 2026 08:20am
- The government is expected to exclude reimbursement expansion-related price cuts from its routine post-listing price cuts, which will take effect next year. The decision reflects concerns that patients could face delays in benefiting from lower drug costs, as well as the administrative complexity of refund procedures.At a public-private consultative meeting on drug pricing reform held on July 22, government and industry representatives discussed measures to improve the standardization of post-listing price cuts.In March, the Health Insurance Policy Deliberation Committee (HIPDC) agreed to align the timing of price cuts under various post-listing management mechanisms to April and October to improve the predictability of reimbursement price reductions.The routine post-listing price cut measure was approved by the HIPDC in March. Price cuts resulting from reimbursement expansions are expected to be excluded.Until now, reimbursement prices have been adjusted whenever a new indication was approved, or reimbursement coverage was expanded. The government sought to reduce these post-listing price cuts to two scheduled adjustments each year—in the first and second half of the year—to improve predictability for the industry.Under the original proposal, reimbursement expansions themselves would still take effect immediately. However, the resulting reimbursement price reduction was set to be deferred until the next scheduled adjustment, with pharmaceutical companies later refunding the difference.For example, if reimbursement coverage were expanded in January and the reimbursement price was therefore subject to a reduction, the amount corresponding to the delayed price cut until the April adjustment would be refunded afterward by the pharmaceutical company.However, this approach raised concerns that patients would not immediately benefit from lower drug costs at the time of reimbursement expansion.In addition, implementing the refund mechanism was expected to create a substantial and unnecessary administrative burden for both pharmaceutical companies and the National Health Insurance Service. As a result, the government has largely decided to exclude reimbursement expansion-related price cuts from the routine post-listing price adjustment schedule.Meanwhile, the government had previously planned to abolish actual transaction price-based price cuts after expanding incentives for lower-price drug purchasing but has decided to maintain the current system due to concerns about potential drawbacks. Reimbursement re-evaluations will also continue to be conducted when warranted rather than on an annual basis. The timing of price adjustments under these mechanisms will likely be brought into line with the new routine schedule.
- Product
- Novartis·Boehringer reportedly restructureing sales rights for blockbuster items
- by Kim JiEun Jul 24, 2026 08:20am
- AI-generated imageAs reports emerge that Novartis and Boehringer Ingelheim are pursuing business reorganizations to transfer the sales rights of major products in South Korea to local pharmaceutical companies or change their distribution partners, the pharmaceutical distribution industry is closely monitoring.It is reported that new distributors have been selected for certain products, or that negotiations are in their final stages, with the expectation that sales rights will transition sequentially as current contracts expire. However, the companies have yet to issue official statements.According to distribution industry sources on July 23, Novartis is reviewing measures to reorganize its sales strategies for several prescription drugs.Following its co-promotion of Gleevec with Yuhan, co-promotion models with domestic companies are also reportedly under consideration for products such as the lung cancer treatment Tasigna, the immune thrombocytopenia treatment Revolade, and the iron overload treatment Exjade.Recently, Novartis also signed a partnership agreement transferring the promotional and supply rights for its cardiovascular products, Diovan and Exforge, to DKSH Korea.Multiple industry sources said, "We understand that directions have already been set to some extent for certain items, while various candidate companies are being reviewed for the remaining products."It is being reported in the industry that Boehringer Ingelheim is pursuing a similar reorganization of its sales strategy.Industry reports indicate that for some products currently co-promoted with Yuhan, discussions are underway to transfer sales rights to other distributors upon contract expiration, with direction already effectively finalized for certain items.In particular, new distributors have reportedly been selected for around five items, including the inhaler Atrovent, and it has been raised that some items currently distributed by Yuhan will also undergo sequential distributor changes as contracts expire.In some quarters, the possibility of structural changes in sales for the diabetes treatment Jardiance is also being discussed.Wholesalers "closely monitoring margin changes"... Pharmacies "return issues could recur"The wholesale industry is reacting most sensitively to these rumored transfers of sales rights. This is because a change in distributor frequently accompanies changes not only to transaction terms, but also to margin structures and return criteria.Given that Novartis and Boehringer Ingelheim hold numerous high-volume products in the domestic market, business partners are closely monitoring potential changes in contract terms.Pharmacies are also expected to face difficulties in managing existing inventory if suppliers change. In the past, pharmacies experienced confusion during sales rights transitions as outgoing and incoming distributors repeatedly shifted responsibility for inventory returns to each other.However, industry analysts suggest that since most of the items involved are high-volume, frequently prescribed products with fast inventory turnover at pharmacies, the actual impact on the field could be limited.The industry views this rights reorganization as having a significant impact on distribution structures and commercial terms beyond a simple change of sales partners. As both Novartis and Boehringer Ingelheim hold substantial market share in major products in the domestic prescription market, wholesalers and pharmacies alike are closely watching for official announcements and contract progress.An industry insider stated, "When sales rights change, the distribution structure ultimately changes as well. The most sensitive aspects are margins and commercial terms from the wholesalers' perspective, and existing inventory management and return standards from the pharmacies' perspective. Although official announcements have not been made yet, the entire industry is keeping a close eye on these developments because the products involved hold such significant influence."Another industry official anticipated, "Because these products have high turnover rates, major chaos at pharmacies may not result. However, if inventory return standards or supply criteria change during the transfer of rights, both distributors and pharmacies could be considerably affected, making the final contract details crucial."