- LOGIN
- MemberShip
- 2026-06-10 16:16:04
- Policy
- Merck’s two new rare disease drugs receive GIFT designation
- by Lee, Tak-Sun May 15, 2026 02:44pm
- AI-generated imageThe Ministry of Food and Drug Safety (MFDS) is speeding up domestic approval timelines by designating two rare disease treatments from Merck as subjects for Korea’s Global Innovative products on Fast Track (GIFT) program.The MFDS announced that it designated Merck’s desmoid tumor treatment ‘Ogsiveo Tab’ and tenosynovial giant cell tumor (TGCT) treatment ‘Pimicotinib Cap’ as the 70th and 71st GIFT products, respectively. This designation is a measure to support the expedited approval of new drugs from innovative pharmaceutical companies that either have no existing treatment options or have demonstrated improved efficacy and safety.Ogsiveo Tab (nirogacestat hydrobromide), which was designated as a GIFT product on April 21, is a treatment for adult patients with desmoid tumors requiring systemic therapy. The drug suppresses tumor growth by inhibiting gamma secretase (GS) and blocking Notch signaling pathways.The drug has already received approval from the US FDA (November 2023) and the European EMA (August 2025), and was designated as an orphan drug in Korea on February 24, 2026. The MFDS selected it for expedited review based on the lack of existing treatment options.Then, on April 27, “Pimicotinib Cap (pimicotinib hydrochloride monohydrate),” a treatment for tenosynovial giant cell tumor(TGCT), was added to the GIFT list. The treatment works by selectively inhibiting the colony-stimulating factor-1 receptor (CSF-1R) to block disease progression.Pimicotinib is currently under development with FDA Fast Track and Breakthrough Therapy Designation (BTD) status in the United States, but has not yet received full approval in any global market. MFDS acknowledged the drug’s potential efficacy improvements and decided to manage it as a fast-track review product.The two products designated under GIFT will benefit from review periods shortened by approximately 25% compared to standard reviews. In addition, a rolling review of prepared materials and customized consultations with professional reviewers prior to the submission of the marketing authorization application will be provided, which is expected to significantly accelerate the timeline for the drug’s introduction in Korea.An MFDS official stated, “The exact indications and efficacy will be finalized after reviewing the submitted data. We will spare no effort in the review process to ensure that patients with intractable rare diseases can be provided with new treatment opportunities as quickly as possible.”
- Company
- GC Biopharma signs biopharma manufacturing MOU with Merck
- by Choi Da Eun May 15, 2026 02:43pm
- GC Biopharma is joining hands with global science and technology company Merck Life Science to strengthen its competitiveness in biopharmaceutical manufacturing. Through the partnership, the companies aim to secure stable supplies of key raw and subsidiary materials while improving manufacturing process efficiency to strengthen global market responsiveness.GC Biopharma announced on the 14th that it signed a strategic memorandum of understanding (MOU) with Merck Life Science for cooperation in biopharmaceutical development and GMP manufacturing processes.The signing ceremony was held at Merck Korea headquarters in Gangnam, Seoul. Major officials from both companies attended, including Young-im Kim, head of Merck Life Science Process Solutions Business, and Woong Shin, head of Quality Management at GC Biopharma.Through this agreement, GC Biopharma plans to strengthen cooperation on the supply of major raw and subsidiary materials needed for biopharmaceutical production and establish a collaborative framework to improve manufacturing efficiency and supply stability.In particular, GC Biopharma expects to secure a stable production base for major global products, such as its plasma-derived therapy Alyglo and the Hunter syndrome treatment Hunterase, both launched in the U.S. market, and respond more flexibly to changes in global demand.Merck plans to provide a collaborative framework covering the entire production process, from raw material procurement to process technology support. In particular, the companies aim to proactively manage supply chain risks that may arise during the manufacturing process by stably supplying product batches that meet strict internal quality control standards.The two companies also plan to operate a regular technical and process consultation body to strengthen technical cooperation for manufacturing process efficiency. Through this, they intend to share the latest production technologies and process know-how to enhance quality competitiveness, while continuously seeking joint R&D opportunities to expand their cooperative relationship.Woong Shin, Head of Quality Management at GC Biopharma, stated, “Through close technical cooperation and process optimization, we will work to minimize risks across the entire manufacturing process and build a production system with global-level quality competitiveness.”Young-im Kim, head of Merck Life Science Process Solutions Business, said, “Through this collaboration, we will work to support an optimal manufacturing environment so that GC Biopharma’s key therapeutic products can be supplied stably.”
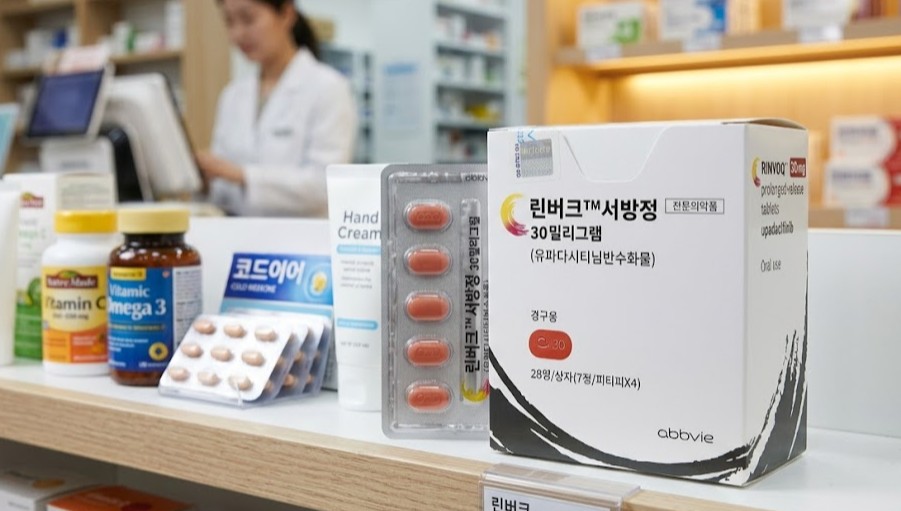
- Company
- Rinvoq surpasses ₩100B in quarterly prescriptions
- by Kim, Jin-Gu May 15, 2026 02:43pm
- Product photo of Rinvoq Extended-release tabletAbbVie’s Janus kinase (JAK) inhibitor autoimmune disease treatment, ‘Rinvoq (upadacitinib),’ has strengthened its leading position by surpassing KRW 10 billion in quarterly prescriptions. Competing products such as ‘Olumiant (baricitinib),’ ‘Xeljanz (tofacitinib),’ and ‘Cibinqo (abrocitinib)’ showed sluggish performance.The JAK inhibitor market is expected to face even fiercer competition. This is due to the addition of new products such as Pfizer’s ‘Litfulo (ritlecitinib)’ and LEO Pharma’s ‘Anzupgo Cream (delgocitinib),’ as well as the launch of generics following the expiration of Xeljanz’s substance patent late last year.Rinvoq strengthens monopoly in JAK inhibitor market through expanded indicationsAccording to the pharmaceutical market research firm UBIST, on the 13th, Rinvoq's outpatient prescription volume in the first quarter was KRW 10.4 billion. This is a 32% increase compared to the KRW 7.8 billion recorded in the first quarter of last year. This marks the first time Rinvoq’s quarterly prescription performance has exceeded KRW 10 billion.Rinvoq received domestic approval in June 2020. Among JAK inhibitors, it was released third following Xeljanz and Olumiant, but it rapidly expanded its influence. Since the fourth quarter of 2023, Rinvoq has risen to the top of the market. Since then, it has been strengthening its monopoly by widening the gap with competing products.In contrast, Rinvoq’s major competitors appear sluggish. Olumiant, the second-ranked product in the market, recorded KRW 4.5 billion in first-quarter prescriptions, a slight decrease compared to KRW 4.6 billion in the first quarter of last year.During the same period, Xeljanz decreased by 21% from KRW 3.5 billion to KRW 2.8 billion KRW. This was influenced by a price reduction following the listing of generics for reimbursement in November last year. The prices of two dosages of Xeljanz tablets were cut by 30%, while the price of Xeljanz XR extended-release tablets was reduced by 23%. ‘Cibinqo,’ which Pfizer launched as a successor to Xeljanz, decreased from KRW 1.6 billion to KRW 1.5 billion.Jyseleca, which entered the market as the fifth JAK inhibitor, is significantly improving its prescribing performance. In the first quarter of this year, it recorded KRW 1.8 billion in prescriptions, a 2.6-fold increase compared to the same period last year.Prescription volume changes for major JAK inhibitors (unit: KRW 100 million, source: UBIST). GREEN-Rinvoq, PINK-Olumiant, BLUE- Xeljanz, LIGHT GREEN- Jyseleca, PURPLE- CibinqoThe aggressive strategy of expanding indications is cited as the background for Rinvoq’s strengthened dominance. Currently, Rinvoq holds six indications ▲rheumatoid arthritis ▲psoriatic arthritis ▲axial spondyloarthritis (ankylosing spondylitis) ▲atopic dermatitis (adults and adolescents) ▲ulcerative colitis ▲Crohn’s disease. It has secured the most indications among JAK inhibitors.In addition, AbbVie recently succeeded in global Phase 3 clinical trials for alopecia areata, signaling the addition of a seventh indication. Besides alopecia areata, AbbVie is also pursuing expansions into subsequent immune disease indications such as vitiligo, hidradenitis suppurativa, systemic lupus erythematosus, and Takayasu arteritis.Its competitor, Xeljanz, has five indications ▲rheumatoid arthritis ▲psoriatic arthritis ▲ankylosing spondylitis ▲ulcerative colitis ▲polyarticular juvenile idiopathic arthritis ▲juvenile psoriatic arthritis. Olumiant holds four indications ▲rheumatoid arthritis ▲atopic dermatitis (adults and children) ▲alopecia areata (adults) ▲juvenile idiopathic arthritis. Cibinqo has only atopic dermatitis (adults and adolescents) as an indication, while Jyseleca has indications for rheumatoid arthritis and ulcerative colitis.Emergence of 6th and 7th JAK inhibitors and Xeljanz generics late last yearFiercer competition in this market is expected in the future with the addition of the sixth and seventh new JAK inhibitor drugs and Xeljanz generics.Recently, ‘Litfulo’ was launched as the sixth JAK inhibitor. This product received domestic approval in September 2024 and was released as a non-reimbursed drug in February of last year. Its indication is alopecia areata in adults and adolescents aged 12 and older. Pfizer now possesses three JAK inhibitors: Xeljanz, Cibinqo, and Litfulo.In September last year, LEO Pharma received approval for ‘Anzupgo Cream,’ the first topical JAK inhibitor in Korea, for the treatment of chronic hand eczema. LEO Pharma officially launched this product in March of this year. It is currently non-reimbursed.In November last year, a large number of generics containing the ingredient tofacitinib entered the market due to the expiration of the Xeljanz patent.A total of 59 companies have received approval for Xeljanz generics, and products from 14 companies, including Daewoong Pharmaceutical, Il-Yang Pharmaceutical, and Chong Kun Dang, are currently listed on the reimbursement list. However, the prescription volume remains still.

- Company
- MSA left unmanaged despite being a rare neurological disorder
- by Son, Hyung Min May 15, 2026 02:43pm
- Concerns are being raised that multiple system atrophy (MSA), a fatal neurodegenerative disease characterized by faster progression and a heavier disability burden than Parkinson’s disease, still remains outside an independent rare disease management framework in Korea.Unlike major overseas countries that operate separate registration and support systems, Korea still lacks even an accurate understanding of the patient population due to mixed disease coding and limited support measures.At the recent “Denmark-Korea Roundtable on MSA Care” held at the Embassy of Denmark in Korea, domestic and international experts discussed the clinical characteristics of MSA, diagnostic limitations, gaps in long-term care, and the need for rare disease designation. Experts on movement disorders from both Korea and Denmark attended the event to share their management systems.“Denmark-Korea Roundtable on MSA Care” event hosted by the Danish Embassy in Korea.Mikael Hemniti Winther, Ambassador of Denmark to Korea, said in his welcoming remarks, “Patients with rare and intractable neurological diseases require not only access to treatment but also long-term care and social support systems. “I look forward to Denmark and Korea sharing clinical experiences and policy models to further develop patient-focused care systems.”MSA is a progressive neurodegenerative disease in which multiple parts of the nervous system, including the basal ganglia, cerebellum, and brainstem, atrophy simultaneously. While it begins with symptoms similar to Parkinson’s disease, such as stiffness and slowed movement, as the disease progresses, autonomic nervous system abnormalities rapidly develop, including orthostatic hypotension, urinary dysfunction, sleep disorders, and respiratory difficulties.As a result, the disease shows limited response to levodopa-based therapies commonly used for Parkinson’s disease, while symptom progression is significantly faster. According to existing data, the average survival period for MSA patients is known to be approximately 6 to 10 years after symptom onset, and it is not uncommon for patients to require walking aids or wheelchairs within a few years of diagnosis.Professor Do-Young Kwon of the Department of Neurology at Korea University Ansan Hospital said, “Although MSA is rare enough to be classified as a rare disease, its clinical and social impact is extremely severe. MSA patients suffer not only from Parkinsonian symptoms but also cerebellar ataxia, autonomic dysfunction, and sleep disorders, and many progress to severe disability within a few years.”Currently, Korean healthcare big data estimates the number of MSA patients at fewer than 3,000 as of 2024. However, in actual clinical practice, many cases are reportedly registered and managed under the Parkinson’s disease code (G20). Academia views this code overlap as one of the major reasons why the true patient population and disease burden are not properly reflected.There are also significant differences in MSA management compared with other countries. The United States operates incentives for MSA drug development under the Orphan Drug Act, while Japan has established patient registration and medical expense support systems through its designated intractable disease framework. Europe likewise operates national-level management systems based on rare disease coding and research networks.In contrast, Korea does not manage MSA within a separate rare disease framework, leading to insufficient support systems tailored to the disease’s characteristics, including long-term rehabilitation, palliative care, and sleep and respiratory management.Rapid progression and complex symptoms…limits of treating MSA the same as Parkinson’s diseaseProfessor Sooyeon Yoo, Seoul Medical Center, Department of NeurologyProfessor Sooyeon Yoo of the Department of Neurology at Seoul Medical Center emphasized that diagnosing MSA itself is not easy.Professor Yoo explained, “MSA often presents symptoms very similar to Parkinson’s disease in the early stages. In some patients, characteristic autonomic nervous system abnormalities or imaging changes only appear after specific symptoms have progressed, so delayed diagnosis is common.”In clinical practice, it reportedly takes an average of 3-4 years to reach an accurate diagnosis. The problem is that during this period, patients may remain in a treatment gap without appropriate rehabilitation or long-term care planning.Professor Yoo said, “MSA patients experience severe declines in quality of life due to sudden fainting, falls, urinary dysfunction, and impaired temperature regulation. It places a significant psychological and financial burden not only on patients but also on their caregivers.”She added, “Since there is currently no fundamental treatment capable of slowing disease progression, it is important to establish support systems that integrate rehabilitation, respiratory care, sleep management, and palliative care over the long term. We need a structure within the public healthcare system that ensures patients receive continuous care.”Professor Jinyoung Youn of the Department of Neurology at Samsung Medical Center also pointed out the limitations of the current system, which manages MSA at the same level as Parkinson’s disease.Professor Jinyoung Youn, Department of Neurology, Samsung Medical Center“Many Parkinson’s disease patients are able to maintain daily life for long periods through medication management, MSA involves a much more rapid decline in physical function. There are currently no medications available to improve cerebellar symptoms.”He further explained that because MSA presents with a complex combination of symptoms, a single-department approach has clear limitations. In fact, some medications may alleviate Parkinson’s symptoms but exacerbate others, such as orthostatic hypotension or constipation.Professor Youn said, “It is common for drugs used to control one symptom to aggravate another. A multidisciplinary approach involving not only neurology but also urology, pulmonology, rehabilitation medicine, and sleep medicine is essential.”In particular, he pointed out that a significant portion of the rehabilitation, sleep therapy, respiratory support, and long-term care services required by MSA patients are still not adequately covered under the current system.Professor Youn noted, “MSA patients often develop the disease at relatively younger ages and tend to preserve cognitive function comparatively well. As a result, many do not fit well within existing long-term care facilities or dementia-centered caregiving systems.”The Danish case stood in contrast to Korea’s current reality.Professor Anne-Mette Hejl of Bispebjerg-Frederiksberg Hospital in Denmark stated, “In Denmark, neurologists, specialized nurses, physical and occupational therapists, and neuropsychology experts collaboratively manage MSA patients. Depending on the patient’s condition, services are linked to telemedicine, home care, and hospice care.”“Denmark-Korea Roundtable on MSA Care” event hosted by the Danish Embassy in Korea.In practice, Denmark operates a system that reduces caregiving burdens for patients and families through follow-up observations every 3 months, extended consultations, and connections to home-based services. When hospital visits become difficult, patients are transitioned to telemedicine and community-based care systems.To date, there are no treatments available that can fundamentally slow or halt the progression of MSA itself. Consequently, experts generally agree that the importance of early diagnosis, rehabilitation, respiratory and sleep management, and long-term care systems is becoming increasingly prominent.Meanwhile, global pharmaceutical companies continue to develop therapies aimed at suppressing MSA progression. Lundbeck’s “Amlenetug” has entered global phase 3 clinical trials, while Teva’s “Emrusolmin,” BioArctic’s “Exidavnemab,” and Alterity Therapeutics’ “ATH434” are also in clinical development stages.Experts believe that if disease-modifying therapies become available in the future, the importance of early diagnosis and national patient registration systems will grow even further.

- Opinion
- [Reporter's View] The dark side of improved diabetes med convenience
- by Son, Hyung Min May 14, 2026 09:28am
- "I lost OOkg with Wegovy," "My appetite completely vanished after taking Munjaro."Personal stories about obesity treatments are now easily seen on social media platforms like YouTube. Content detailing how much weight was lost, what the side effects were like, and which drugs are more effective is being consumed just like any other everyday lifestyle content.Most experts emphasize that obesity is not a simple issue of body shape but a chronic disease that requires medical treatment. In reality, obesity is linked to various metabolic disorders such as diabetes, cardiovascular disease, and fatty liver, necessitating a therapeutic approach that considers Body Mass Index (BMI) and the presence of comorbid conditions.However, the reality is that the perception of obesity treatments is increasingly shifting toward 'weight-loss drugs' rather than treatments for a disease.The pharmaceutical industry's emphasis on improving medication convenience has also played a significant role in this trend.The rapid expansion of the GLP-1 class obesity treatment market is due to the convenience of once-weekly administration. Compared to past treatments that required daily oral intake or injections, the burden of use has been significantly lowered, leading to expanded patient access and market growth.Improvements in medication convenience are evaluated as meaningful changes in terms of patient accessibility and treatment persistence. Indeed, reducing medication burden in chronic disease management improves treatment adherence and patient quality of life. Accordingly, the pharmaceutical industry continues to develop drugs that maintain efficacy for longer periods with fewer administrations.Currently, the pharmaceutical industry is accelerating the development of various forms of obesity drugs, including oral pills, once-monthly injections, and patches. In a situation where even injections are accepted without much hesitation, the threshold for use is likely to drop even further once new drugs with drastically improved convenience emerge.The problem is that improved medication convenience does not carry the same meaning as improved medication compliance.Medication compliance is a concept closer to a patient who consistently maintains therapy according to the proper use and dosage. However, in the current obesity treatment market, 'how easily it can be used' is emphasized first, while discussions on who should use it and how are relatively lacking.In the online market, health-functional foods and overseas direct-purchase products, some with names similar to those of GLP-1 agents, are spreading rapidly, even though they are unrelated. Some of these products are consumed for weight loss purposes despite lacking sufficient medical validation or safety assessments.Of course, there is no reason to deny the clinical value of obesity treatments themselves. GLP-1 class treatments are changing the paradigm of obesity management by accumulating diverse data, including reductions in cardiovascular disease risk beyond weight loss. They are undoubtedly an important option for patients who require treatment.As treatments become more popular, what needs to grow alongside the consumption craze is a clear understanding of medical treatment. As convenience and accessibility increase, social standards regarding prescription criteria and medical necessity must become clearer. At the very least, there is a need to guard against obesity treatments becoming firmly established as merely 'diet shots that anyone can easily administer.'

- Policy
- New drug review timeline cut from 295 to 240 days
- by Lee, Tak-Sun May 14, 2026 09:28am
- As the Ministry of Food and Drug Safety (MFDS) moves to shorten new drug approval review timelines from 295 days to 240 days while strengthening communication with companies. The agency will introduce face-to-face meetings between companies and reviewers prior to the application stage, as well as a “checklist” for companies to self-review their data, aiming to reduce the total approval period by nearly 2 months compared to the previous process.The MFDS announced that it has prepared a revised draft of the “New Drug Product Approval and Review Procedures (Civil Servant Guidelines),” which is currently undergoing a public comment period, and plans to fully implement it starting October 1. This revision, coming approximately one and a half years after the guidelines were established in December 2024, was pursued to maximize the predictability of new drug approvals.From post-submission to pre-submission… introduction of advance face-to-face meetingsThe most significant change is the introduction of ‘pre-NDA meetings.’ Under the original 2024 version, a dedicated team was formed and the review began within 10 days of receiving the application; however, the revised version requires the company and the MFDS to begin discussions 3 months prior to application submission.When a company requests a face-to-face meeting, a dedicated team is formed immediately, and through at least 2 meetings, any deficiencies in the data will be identified in advance. This is expected to serve as a key mechanism to accelerate the entire process by reducing the time spent on ‘requests for additional information,’ which frequently occur during the official review stage.AI-generated graphic imageIntroducing a ‘checklist’ to prevent ‘insufficient data’ at the sourceThe ‘checklist’ system, which requires companies to self-verify the completeness of their submitted data, is another key change in this amendment. Previously, the MFDS would notify companies of required corrections on a case-by-case basis after receiving the data; now, companies must complete a self-inspection using a detailed checklist starting from the face-to-face meeting stage prior to application.Products that undergo this procedure will see a significant reduction in errors or omissions in their documentation, resulting in the “preliminary review” period, conducted immediately after submission, being shortened from the previous 7 days to within 3 days.Approval timeline shortened by 55 days… “295 Days → 240 Days”Through these strengthened communication systems, the Ministry of Food and Drug Safety (MFDS) has set the target approval period for new drugs at 240 days, down from the previous 295 days. This represents a reduction of approximately 55 days compared to the original regulations.In addition, the govenrment has maximized review efficiency by codifying a ‘rolling review’ procedure for frequent exchange of opinions during the review process and by moving the GCP (Good Clinical Practice) site inspection, which previously took place after the first round of supplementary submissions, to the early stages of the review (within 60 to 120 days after submission).An MFDS official stated, “This revision goes beyond simply shortening the timeline; it institutionalizes ‘pre-submission communication’ and ‘self-assessment by companies’ to overcome the limitations identified during the operation of the original version. Once the new procedures take effect this coming October, the speed at which innovative new drugs enter the market, both domestically and internationally, will increase dramatically.”The industry’s response has also been favorable. An official from the Korea Biomedicine Industry Association said, “The introduction of the checklist will help ensure that more comprehensive data is submitted when applying for new drug approval and will likely reduce the need for supplementary data in the future. This is interpreted as a positive gesture where the MFDS and companies join hands to successfully complete the product approval process.”

- Company
- Domestic CAR-T Limcato speeds toward reimbursement
- by Hwang, byoung woo May 14, 2026 09:28am
- With Korea’s first domestically developed CAR-T therapy Limcato (anbalcabtagene-autoleuce) receiving regulatory approval, attention is now focused on the timing of its reimbursement.Curocell expects to accelerate market entry through the pilot program that allows parrallel operation of approval, reimbursement evaluation, and price negotiation. With the fast-track system in place, commercialization may be possible as early as the second half of the year.However, some observers note that it remains to be seen whether the reimbursement listing will proceed strictly according to schedule. While Limcato holds symbolic significance as the first domestically developed CAR-T therapy, it is still an ultra-high-cost, one-time treatment costing hundreds of millions of won.This implies that factors such as risk-sharing, performance-based post-marketing management, and reimbursement conditions, in addition to the drug price, could serve as variables in the negotiation process.Ultimately, the key focus regarding Limcato’s reimbursement is shifting from “whether it will be reimbursed” to “under what conditions and how quickly it will be listed.”While the likelihood of reimbursement itself is viewed relatively positively, analysis suggests that the actual speed of commercialization may vary depending on the terms of the negotiations.Reimbursement prospects are optimistic… Speed expected to rise via parallel trackLimcato is an autologous CD19-targeted CAR-T therapy developed by Curocell. Its approved indications are for the treatment of adult patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) and primary mediastinal large B-cell lymphoma following two or more prior line of systemic therapy.The Ministry of Food and Drug Safety (MFDS) granted marketing authorization for Limcato on the 29th of last month. This marks the first instance of a CAR-T therapy developed by a domestic company receiving approval.Previously, the MFDS designated the drug as a “Bio-Challenger” candidate and the 33rd entry in the “Global Innovative Product on Fast-Track (GIFT)” system, providing tailored consultation and expedited review from the early stages of development.The outlook for reimbursement is quite promising. Limcato has been selected as a drug for the Ministry of Health and Welfare’s “Pilot Project for Parallel Application for Approval, Reimbursement Evaluation, and Drug Price Negotiation.”Previously, the process could take over 300 days, including 120 days for MFDS approval, 150 days for HIRA reimbursement evaluation, and 60 days for NHIS drug price negotiations, but the pilot project aims to shorten this timeline.The company also maintains a positive outlook. Regarding drug price negotiations, the company has mentioned the possibility of an adjustment to a level that is the same as or slightly lower than the prices of existing CAR-T therapies, and therefore does not view the drug price itself as a factor that will cause significant delays.In Korea, Novartis’ Kymriah has already established a precedent for CAR-T reimbursement, making this scenario more feasible.Last January, the National Health Insurance Review and Assessment Service (HIRA) established reimbursement criteria for Gilead Sciences’ Yescarta for the indication of “treatment of adult patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) and primary mediastinal B-cell lymphoma (PMBCL) following two or more prior systemic therapies.”Although Limcato is a late entrant, it benefits from accumulated experience in prior CAR-T evaluations and negotiations.However, the industry notes that reimbursement likelihood and negotiation difficulty are separate issues. Even if the likelihood of entering the reimbursement system is high, given the nature of ultra-high-cost one-time treatments, there may be significant debate surrounding the conditions for listing.Conditions, not price, may be the key variable… focus on risk-sharing discussionsPricing is an important starting point for Limcato. Since Kymriah is already reimbursed, the CAR-T therapy’s price is expected to serve as a benchmark.If Limcato adopts a relatively lower pricing strategy, this could strengthen its case for reimbursement. Its domestic manufacturing base also adds positive policy value.However, industry experts suggest that risk-sharing and post-management conditions may be more critical than price itself.CAR-T is an ultra-high-cost therapy that is expected to produce therapeutic effects with a single administration. If a sufficient response does not occur after administration, or if relapse or death occurs within a certain period, it will be crucial to determine how to link the financial burden on the national health insurance system to treatment outcomes.According to Market Access (MA) experts, the likelihood of Limcato being covered by insurance is considered relatively high. This is because there are precedents for the coverage of existing CAR-T therapies, and if the company proposes a price that is equal to or lower than that of existing treatments, there is a strong possibility that the Health Insurance Review and Assessment Service (HIRA) and the National Health Insurance Service (NHIS) will positively consider the necessity of listing the drug.However, the pharmaceutical industry views that, given the high likelihood of Limcato being discussed within the category of ultra-high-cost one-shot therapies, conditions such as performance-based reimbursement, patient-specific tracking, efficacy evaluation over a set period, and submission of post-treatment management data may be attached.This is assessed not so much as a factor that makes reimbursement itself difficult, but rather as a variable that influences the duration of negotiations and the burden of actual commercialization preparations.In particular, Limcato is the first domestically developed CAR-T therapy to be commercialized directly by a Korean biotech company. Compared to multinational pharmaceutical companies, the organizational burden may be relatively greater in areas such as managing risk-sharing agreements, establishing collateral, managing refunds, responding to hospitals, the National Health Insurance Service (NHIS), and the Health Insurance Review and Assessment Service (HIRA), and managing long-term follow-up data.The industry is considering two scenarios regarding this point.The first scenario is if the company accepts the drug price and post-marketing management conditions relatively quickly. In this case, leveraging the intent of the pilot program, which combines approval, evaluation, and negotiation, it appears possible to secure reimbursement listing and achieve commercialization in the second half of the year.If Limcato secures price competitiveness compared to existing CAR-T therapies and negotiations proceed without major disagreements regarding post-marketing management conditions, the speed of market entry could accelerate.The second scenario is if negotiations over risk-sharing conditions drag on. Even if the drug price can be adjusted relative to existing treatments, discussions regarding reimbursement conditions, long-term follow-up criteria, and performance evaluation methods could take a long time.In this case, rather than the reimbursement listing itself falling through, it is more likely that the timing of the listing and the actual pace of prescription expansion will be delayed.The initial key point to watch is the timing of the submission to the Cancer Drug Deliberation Committee. Even if the drug enters the parallel approval, evaluation, and negotiation track, it must still undergo deliberation by the Cancer Drug Deliberation Committee. The industry is closely watching whether it will be submitted in May.Some observers believe there will be no Cancer Drug Deliberation Committee meeting scheduled for June, so the outline of the reimbursement schedule for the second half of the year is expected to become clearer depending on the timing of the agenda’s submission.Limcato clinical results (Excerpt from Curocell IR Materials) Anbal-cel demonstrates superior treatmnet effect and lower side effect compared with 3 FDA-approved marketed CAR-T therapiesSupply speed is a strength… Differentiation through domestic manufacturing infrastructureIn the commercialization phase following reimbursement, Limcato’s supply competitiveness is expected to emerge as a key differentiator.Existing CAR-T therapies followed a structure where patient cells were collected, sent to overseas manufacturing facilities, and the finished product was then imported back into the country.In this process, manufacturing, logistics, and customs clearance procedures overlapped, requiring a certain amount of time before treatment could begin. Particularly for patients with rapidly progressing terminal blood cancers, the waiting period itself has been cited as a barrier to treatment accessibility.In contrast, Curocell has established a system for domestic manufacturing and supply based on its CAR-T-dedicated GMP production facility in Daejeon. In fact, the company highlights the ability of Limcato to reduce waiting times through this domestic production and supply system as a key strength.Curocell has also established “CuroLink,” an integrated solution for commercial operations. CuroLink is a cell therapy supply management solution that links information in real time between hospitals, manufacturing sites, and logistics providers. It is structured to track and manage patient-specific information from prescription through leukocyte collection, cell manufacturing, shipment, and administration.Curocell also believes that its domestic production base will help expand patient access.A Curocell official stated, “Since Limcato is manufactured at domestic GMP facilities, we can promptly respond to supply requests even from regional hospitals. Once a hospital decides to prescribe the drug after it is listed for reimbursement, the product supply process can be carried out through the system prepared by the company.”

- Company
- Medipost's Cartistem Phase 3 clinical trial succeeds in Japan
- by Cha, Ji-Hyun May 14, 2026 09:28am
- Medipost has met all dual primary endpoints in its Japanese Phase 3 clinical trial for Cartistem, a stem cell therapy for knee osteoarthritis. By securing statistical significance in both subjective symptom improvement and arthroscopic cartilage regeneration compared to the active control group, the company’s efforts for commercialization in Japan and the advancement of its U.S. Phase 3 trials are expected to speed up.On the 13th, Medipost held a press conference at the Four Seasons Hotel in Jongno-gu, Seoul, to disclose the major results of the Japanese Phase 3 trial for Cartistem and its future global development strategy. Key attendees included Medipost CEO Wonil Oh, Global Business Head and CEO of Medipost K.K. (Japan) and co-CEO of Medipost Inc. (USA) Seung Jin Lee, and Vice President Hoon Sik Cho.Medipost CEO Wonil OhCEO Wonil Oh remarked, "This achievement is a major achievement of Medipost's journey since its founding, driven by the goal of improving patients' quality of life through stem cell therapy innovation," and added, "We will achieve entry into the Japanese market for Cartistem and complete the ongoing U.S. Phase 3 trial to emerge as a global leader in the stem cell industry."Cartistem is an allogeneic umbilical cord blood-derived mesenchymal stem cell therapy developed by Medipost. It is a treatment aimed at regenerating damaged cartilage and improving pain and function in patients with knee cartilage defects caused by degenerative changes or repeated trauma. Since receiving marketing authorization from the Ministry of Food and Drug Safety (MFDS) in 2012, it has accumulated over 10 years of prescription experience. Cartistem recorded sales of KRW 19.5 billion last year.Medipost conducted Japanese clinical trials to overcome domestic market growth limitations and secure its first overseas commercial hub. The Japanese Phase 3 trial was designed as a pivotal study for Japanese marketing authorization. Based on its experience in authorization and commercialization, as well as existing clinical data accumulated in Korea since 2012, Medipost was able to skip Phase 1 and 2 trials in Japan and enter directly into Phase 3.The trial was a randomized, active-controlled comparative study conducted at 13 medical institutions in Japan involving 130 patients with knee osteoarthritis. The actual administration and surgery groups consisted of 59 patients in the Cartistem group and 61 in the hyaluronic acid (HA) group. Patients were followed for 52 weeks after administration to observe efficacy and safety.The control group received a hyaluronic acid injection, which is the standard of care for patients with knee osteoarthritis in Japan. Global Business Head Seung Jin Lee explained, "In the process of discussing the clinical design with the Japanese Pharmaceuticals and Medical Devices Agency (PMDA), we reflected the fact that Japanese patients generally receive HA injections for knee osteoarthritis treatment and set that product as the control."The primary subjects were patients with knee osteoarthritis aged 20 to 80. The radiographic severity of osteoarthritis corresponded to K&L grades 2 to 3, and the degree of cartilage damage corresponded to ICRS grades 3 to 4. The size of the cartilage defect ranged from approximately 2 to 9 square centimeters, and the trial included patients with a pain score of 40 or higher on a 100mm Visual Analog Scale (VAS) and a Body Mass Index (BMI) of less than 35. Furthermore, the study targeted patients who had not shown sufficient symptom improvement despite receiving existing treatments, such as exercise therapy, physical therapy, or hyaluronic acid injections, for at least 3 months.According to the presentation, Cartistem met both dual primary endpoints in the Japanese Phase 3 trial. The dual primary endpoints were ▲the change in the WOMAC total score ▲the improvement rate of one or more ICRS grades. WOMAC is a patient-reported outcome that evaluates knee pain, functionality, and stiffness as perceived by the patient. ICRS is a structural evaluation metric used to confirm whether the degree of cartilage damage has actually improved through arthroscopy. The design required both metrics to be met for the trial to be considered a success.Lee explained, "Since Cartistem is administered via surgery and HA via injection, patients inherently know which treatment they received, which can allow for a placebo effect in patient-reported metrics," and added, "To compensate for this, we included the ICRS evaluation, which directly checks the cartilage state via arthroscopy at the 52-week mark, as a co-primary endpoint. To ensure the treatment group information remained undisclosed, evaluations were conducted by a separate central adjudication committee after blinding the data."Medipost CEO Wonil Oh, Global Business Head and CEO of Medipost K.K. (Japan) and co-CEO of Medipost Inc. (USA) Seung Jin Lee, and Vice President Hoon Sik ChoClinical results showed that Cartistem demonstrated statistically significant superiority in changes in the WOMAC total score compared with the HA group. The WOMAC p-value was reported as <0.0001. Cartistem also showed a significant improvement over the active control group in the rate of improvement in one or more ICRS grades, with an ICRS p-value of 0.0002.Consistent results were also confirmed in the secondary endpoints. The VAS for pain level and the IKDC and KOOS for knee functionality all recorded p-values of less than 0.0001. Significant improvements were also observed across all WOMAC sub-items, including pain, functionality, and stiffness."This Japanese Phase 3 study was designed so that it had to meet both dual primary endpoints," Lee said. "It is highly significant in that we confirmed not only the improvement in pain and function felt by the patient but also the regeneration of cartilage via arthroscopy," and added, "In the U.S. or Europe, a design that performs arthroscopy on patients again is not easy due to the high patient burden and ethical considerations, as it involves anesthesia and invasive procedures. Therefore, the ICRS data secured in the Japanese Phase 3 will be highly valuable data to be utilized overseas."Medipost plans to apply for Japanese marketing authorization in the second half of this year based on these clinical results. Given the PMDA review period, the company aims to obtain approval by the end of 2027. It plans to pursue a launch in the Japanese market, followed by subsequent processes for price setting and insurance reimbursement.The company expressed strong confidence regarding the possibility of Japanese marketing authorization. Lee stated, "Due to the nature of cell and gene therapies, CMC (which refers to manufacturing process management and quality consistency), inspections of our GMP facility in Guro, Korea, and our local production partner's facility in Japan, remain." However, Lee said, "After reviewing these results with external experts, including advisory panels and former PMDA officials, we received the opinion that with clinical results indicate there should be no major issues in gaining marketing authorization."The local partner, Teikoku Pharma, will handle sales in Japan. Previously, Medipost signed an exclusive distribution agreement for Cartistem in Japan with Teikoku Pharma in December last year. The company has already received a non-refundable upfront payment of USD 8 million, with an additional USD 10 million payable upon marketing authorization. Lee explained, "The marketing authorization holder will be Medipost K.K., the Japanese subsidiary of Medipost, and Medipost will also manage the responsibility for manufacturing and shipping. Teikoku Pharma will be responsible for transportation, sales, and marketing within Japan."Considering the price-setting and insurance reimbursement procedures following Japanese marketing authorization, Medipost is also conducting a Real-World Evidence (RWE) study in South Korea. The company explained that it has secured data from approximately 550 patients who underwent Cartistem procedures at 13 Korean medical institutions, with an average follow-up period of about 6.9 years."Since Cartistem received marketing authorization in Korea in 2012, approximately 36,700 patients have undergone surgery as of the end of last month," Lee emphasized. "Among them, we are collecting real-world evidence data targeting patients who have passed at least three years since their procedure."Lee continued, "The endpoint we consider most important is the rate of conversion to total knee artificial joints. From the data of 550 patients secured so far, we have identified that less than 1% of patients have converted to artificial joints." He added that detailed information would be disclosed at the end of the year. The company intends to use this data as reference material for the Japanese marketing authorization application and as evidence during the price-setting and insurance reimbursement negotiations with the Ministry of Health, Labor and Welfare after approval.Lee noted, "In Japan, the process of price setting and insurance reimbursement with the Ministry of Health, Labour and Welfare proceeds after marketing authorization. How many years a patient maintains the effect after receiving Cartistem, and the rate at which they transition to other treatments or artificial joint surgery, can be vital data for economic evaluations," and stated, "This real-world evidence study will serve as an important basis not only for Japan but also for discussions with insurance companies in the U.S. after obtaining approval there."Regarding the company's U.S. Phase 3 clinical trial, patient enrollment is currently underway following the U.S. Food and Drug Administration (FDA) approval of the IND application earlier this year. The company plans to use the results of the U.S. Phase 3 trial, along with the Japanese Phase 3 data and Korean RWE, as materials for the U.S. marketing authorization application.

- Company
- Myelofibrosis drug Omjjara nears reimb listing on 2nd attempt
- by Eo, Yun-Ho May 14, 2026 09:28am
- The novel myelofibrosis treatment Omjjara may soon be listed for reimbursement in Korea.According to industry sources, GSK Korea and the National Health Insurance Service (NHIS) have recently finalized price negotiations for the company’s myelofibrosis treatment Omjjara (momelotinib). Barring any unexpected developments, reimbursement is expected to begin as early as June.Omjjara passed the Cancer Drug Deliberation Committee of the Health Insurance Review and Assessment Service (HIRA) in March last year. However, the listing process was halted due to disagreements between GSK and HIRA over the selection of comparator drugs for pricing calculation during the evaluation process.GSK later supplemented its data and passed the first Drug Reimbursement Evaluation Committee of the year, entering price negotiations in February.The specific indication for listing is “the treatment of intermediate- or high-risk myelofibrosis in adults with anemia.”Omjjara has a triple mechanism of action, inhibiting JAK1, JAK2, and ACVR1 (activin A receptor type 1). In myelofibrosis, inhibition of JAK1 and JAK2 improves systemic symptoms and reduces splenomegaly, while ACVR1 inhibition reduces hepcidin expression, thereby alleviating anemia.Anemia management has been one of the key unmet needs in myelofibrosis treatment. Anemia, which increases dependence on blood transfusions, is more than just the dizziness commonly associated with it. Depending on its severity, it can lead to a life-threatening condition.Omjjara has been shown in the Phase III SIMPLIFY-1 and MOMENTUM studies to significantly improve key symptoms, such as splenomegaly, and reduce transfusion dependency in patients with myelofibrosis and anemia, regardless of prior JAK inhibitor treatment history.In the SIMPLIFY-1 study, which evaluated Omjjara as a first-line treatment in JAK inhibitor–naïve patients, Omjjara showed non-inferiority to Jakavi (ruxolitinib) in the primary endpoint of spleen volume response at 24 weeks.The proportion of transfusion independence was 66.5% in the Omjjara group compared to 49.3% in the ruxolitinib group, indicating significantly lower transfusion dependency with Omjjara.Professor Seo-yeon Ahn of Chonnam National University Hwasun Hospital said, “While existing JAK inhibitors improve splenomegaly and systemic symptoms, they have limitations such as worsening anemia and increased transfusion dependency. Omjjara has demonstrated significant clinical value in managing anemia, which is closely linked to prognosis in myelofibrosis patients, and its launch in Korea is expected to contribute to improved treatment outcomes and quality of life for more patients.

- Company
- GE, refining fatty liver quantification…expanding ultrasound tech
- by Hwang, byoung woo May 13, 2026 09:10am
- GE Healthcare is expanding its scope of application in the fatty liver quantification and abdominal ultrasound automation markets, showcasing its next-generation ultrasound platform.While ultrasound-based fatty liver quantification technology has been used previously, the key distinction here is that it has been updated to report liver fat content as a percentage and reduce operator variability.As interest in the co-management of fatty liver and metabolic diseases grows alongside weight loss due to the expanded use of obesity treatments recently, the company's strategy is to broaden the clinical role of ultrasound.On the 12th, GE Healthcare Korea unveiled the LOGIQ R5, which applies the next-generation ultrasound platform 'R5.' Dr. Naohisa Kamiyama, Global Manager of Ultrasound New Clinical Applications at GE Healthcare, who led the development of R5, participated in the presentation and focused on the accessibility of ultrasound in liver disease diagnosis and long-term monitoring.Fatty liver assessment, from 'detection' to 'tracking changes'The focus of this conference was on how far the role of ultrasound can be expanded in the evaluation of fatty liver.Dr. Naohisa Kamiyama, Global Manager of Ultrasound New Clinical Applications at GE HealthcareFatty liver is not easy to detect early because early subjective symptoms are not distinct. If management is delayed, it can lead to steatohepatitis, fibrosis, and cirrhosis, so it is important to check the degree of liver fat accumulation and track it over the long term.This means that, beyond simply confirming the presence or absence of fatty liver, it is necessary to quantitatively assess the liver fat content to determine the disease progression stage and treatment direction.In this process, MRI-PDFF is utilized as a major reference test for fatty liver quantification.MRI-PDFF is a method for measuring the fat fraction in liver tissue as a percentage using MRI. While it has strengths in terms of accuracy and reproducibility, there are constraints on cost and accessibility that limit its broad use in screening or repeated follow-up management.Dr. Kamiyama also acknowledged the role of MRI, noting that ultrasound has strengths in screening and repeated examinations.Dr. Kamiyama said, "MRI is the most important standard in fatty liver quantification," but added, "In areas where repeated checks are necessary, such as health checkups, ultrasound may be more suitable in terms of cost and accessibility."In particular, the spread of obesity treatments can align with this demand for ultrasound-based quantitative evaluation. This is because, as obesity treatments are used in actual clinical settings, interest in changes in liver fat and the co-management of metabolic diseases after weight loss is growing.This is also the background behind GE Healthcare's decision to put fatty liver quantification and abdominal automation functions at the forefront of the LOGIQ R5.The view is that, even if MRI plays a major role in the precision evaluation of high-risk groups, ultrasound can be a tool for more frequent monitoring of patient status in screening and follow-up management.UGFF, Presenting fatty liver status more intuitivelyOne of the most noteworthy technologies in the LOGIQ R5 is 'UGFF (Ultrasound-Guided Fat Fraction),' a fatty liver quantification solution that displays liver fat content as a percentage based on ultrasound.The core is not that it started fatty liver quantification from scratch, but that it refined existing technology in a way that is easy for patients and medical staff to understand. While existing ultrasound-based technology quantifies the degree of fatty liver using attenuation coefficients and the like, UGFF shows the percentage of the liver occupied by fat.LOGIQ R5 product photoPark Do Hyeong, US GI/PC Segment Team Leader at GE Healthcare Korea, said, "In the case of the existing UGAP, the units might have been familiar to medical staff, but it was difficult for patients to understand intuitively." He added, "UGFF is easier to explain to patients in that it shows what percentage of the liver fat occupies."Technologically, it is characterized by the combination of multiple acoustic parameters.UGFF analyzes not only the ultrasound attenuation coefficient but also the integrated backscatter coefficient and signal-to-noise ratio together. When fat accumulates in the liver, the attenuation, brightness, and tissue texture of the ultrasound signal change, and UGFF quantifies the liver fat content by reflecting these changes in a complex manner.Dr. Kamiyama said, "The prevalence of liver disease is high in the Asian region, and the clinical demand for early diagnosis and repeated monitoring is very high." He added, "UGFF is a solution developed by combining multi-acoustic parameters and statistical technology, designed to evaluate liver fat changes more precisely using only ultrasound."Another pillar of LOGIQ R5 is AI-based automation for abdominal ultrasound. This method automatically avoids structures that should be avoided during measurement, such as blood vessels, artifacts, and focal lesions, and adjusts the measurement position during liver fat measurement.Dr. Kamiyama stated, "Ultrasound is significantly affected by the operator and the patient's condition," and added, "We have been improving it in a direction that reduces operator dependence through automatic image optimization and measurement position correction functions."Early launch phase, Expanding clinician awareness is the challengeIn terms of the market, the key for the LOGIQ R5 is whether the advanced fatty liver quantification function can be introduced into medical institutions.GE Healthcare introduced the R5 in Korea in March of this year, following the LOGIQ R4, and is applying it across the LOGIQ series. Some existing equipment must be upgraded from R4 to R5 to use the new functions.In this regard, the company's position is that, since it is still in the early stages of launch, it is necessary to raise awareness and engage in academic communication with medical staff.In addition, as ultrasound-based fatty liver quantification is not the exclusive domain of GE Healthcare, competition is expected to intensify.GE Healthcare is emphasizing an integrated model that combines three parameters as its point of differentiation.YongDuk Kim, CEO of GE Healthcare Korea, stated, "The LOGIQ R5 is a next-generation ultrasound solution developed considering both the increasing number of liver disease patients and the burden of examination for medical staff," and concluded by adding, "GE Healthcare will continue to strive to support the clinical decision-making of medical staff and increase the value of the entire patient diagnosis journey."