- LOGIN
- MemberShip
- 2026-06-12 18:11:50

- Policy
- Demands for expanded reimbursement for NMOSD
- by Jung, Heung-Jun Oct 13, 2025 06:02am
- The demand to improve reimbursement to enhance treatment accessibility for Neuromyelitis Optica Spectrum Disorder (NMOSD) is anticipated to heat up again in this year's parliamentary inspection. During the parliamentary inspection of the Health Insurance Review & Assessment Service (HIRA), on October 17, a NMOSD will attend as a testifier, urging improvements to reimbursement for new drug insurance. NMOSD treatments are gradually receiving reimbursement and expanded criteria. The scope of reimbursement for Roche Korea's Enspryngg (satralizumab), listed for reimbursement in 2023, was expanded in August this year after the symptom relapse criteria had been eased. Uplizna (inebilizumab) recently received conditional reimbursement decision from the Drug Reimbursement Evaluation Committee (DREC) on the 2nd and is awaiting drug price negotiation. AstraZeneca Korea's Soliris (eculizumab) has been covered by reimbursement since April of last year, and Ultomiris (ravulizumab) added the NMOSD indication in July last year but is not yet covered by reimbursement. While access to pharmaceuticals is gradually improving with expanded reimbursement coverage, there are ongoing demands to relax the stringent criteria associated with expensive orphan drugs. In August, a caregiver of an NMOSD patient had requested a lowering of reimbursement hurdles, such as the relapse criteria, through a National Assembly petition. Rep. Seo Mi-hwa of the Democratic Party last month also pointed out the unreasonableness of the new drug reimbursement criteria being conditional on relapse and being preconditioned. Furthermore, there were several arguments that accessibility must be increased for drugs that could prevent relapse. The reimbursement criteria are based on symptom relapse for Enspryngg and Soliris and they also include a conditional clause requiring the administration of MabThera (rituximab) with reimbursement. Since Rep. Seo has requested a NMOSD patient as a testifier for the upcoming parliamentary inspection, more attention is likely to be paid to requests for expanded reimbursement and criteria improvement. Strengthening access to orphan drugs is a key issue that has frequently been raised during the Health and Welfare Committee's parliamentary inspection. Following last year's criticism of the low prio-approval rate for Soliris, the prior-review criteria for its use in Atypical Hemolytic Uremic Syndrome (AHUS) were improved this month.
- Policy
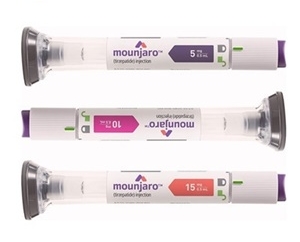
- Ozempic nears reimb approval…next is Mounjaro
- by Jung, Heung-Jun Oct 13, 2025 06:02am
- With Novo Nordisk’s GLP-1 injectable Ozempic (semaglutide) deemed adequate for reimbursement for diabetes, attention is now turning to whether Eli Lilly’s Mounjaro (tirzepatide) will be reviewed next by the Health Insurance Review and Assessment Service (HIRA). At the same time, voices are growing in favor of extending insurance coverage—at least partially—to high-risk obesity treatment, based on body mass index (BMI) criteria. On the 2nd, the Health Insurance Review and Assessment Service's Drug Reimbursement Evaluation Committee recognized the appropriateness of coverage for Ozempic 2mg/1.5mL and 4mg/3mL, used for diabetes patients. Following price negotiations, the final listing procedure will be complete.. Lilly Korea’s Mounjaro, which was being prepared for reimbursement evaluation simultaneously with Ozempic, was not included in the list of drugs submitted to DREC this time. However, Lilly has reportedly been actively pursuing reimbursement for Mounjaro as a diabetes treatment since before its launch, submitting supplementary data. The key question is whether Mounjaro will be submitted to the next DREC meeting while Ozempic undergoes the price negotiation process. Apart from the push for the drugs’ reimbursement as a diabetes treatment, calls for the drugs’ reimbursement as an obesity treatment continue. A reimbursement plan limited to high-risk obesity treatment was mentioned in review materials on GLP-1 reimbursement that Representative Mi-hwa Seo of the Democratic Party of Korea received from the National Assembly Research Service. The review contained an opinion suggesting restricting the target population to patients with severe obesity and those at risk of complications. Furthermore, Representative Seo emphasized the need for public support, citing the higher obesity incidence rate among low-income groups compared to higher-income groups. The Korean Society for the Study of Obesity is also pushing for coverage. At a symposium on health insurance policies for obesity management last September, the society proposed a tiered coverage system based on BMI. Their stance is to prioritize improving treatment access for severe obesity, not cosmetic concerns. However, opinions on introducing coverage remain divided, citing concerns about misuse of obesity drugs and the current state of Korea’s health insurance finances. Furthermore, even if a phased coverage approach based on severity is implemented, challenges remain, including establishing appropriate criteria.
- Company
- Will gastric cancer-targeted therapy Vyloy pass CDDC review?
- by Eo, Yun-Ho Oct 10, 2025 06:06am
- Attention is focused on the second attempt for insurance reimbursement of the gastric cancer-targeted anticancer drug Vyloy. According to industry sources, Astellas Korea’s Vyloy (zolbetuximab), a Claudin 18.2-positive gastric cancer-targeted therapy, is currently coordinating its schedule for review by the Health Insurance Review and Assessment Service’s Cancer Disease Deliberation Committee. The application is expected to be listed on the agenda this month. Vyloy failed to pass CDDC review last February. Astellas resubmitted its reimbursement application in June. Approved in Korea in September last year, Vyloy is the first globally approved Claudin 18.2-targeted treatment, an immunoglobulin monoclonal antibody that binds to Claudin 18.2, a protein expressed and exposed in the stomach. According to the Phase III SPOTLIGHT study that became the basis of Vyloy’s approval, the median progression-free survival (mPFS) of the combination of Vyloy and mFOLFOX6 (oxaliplatin, leucovorin, fluorouracil) was 10.61 months, compared to 8.67 months for the placebo group. The median overall survival (mOS) was also longer—18.23 months versus 15.54 months in the placebo group. In the GLOW study as well, the combination of Vyloy and CAPOX (capecitabine plus oxaliplatin) achieved an mPFS of 8.21 months, reducing the risk of disease progression or death by approximately 31%. However, Vyloy remains non-reimbursed in Korea. It was listed for discussion at the CDDC in February, but failed to establish reimbursement standards. Additionally, a companion diagnostic issue delayed its launch in Korea until March this year. To use Vyloy, patients must be identified as Claudin 18.2-positive, but the companion diagnostic device (CDx) used for this test was under consideration for new medical technology evaluation. To address this, Astellas began an Early Access Program (EAP) even before approval so that patients in need could use the drug in advance. Currently, 51 patients across 10 institutions are enrolled. Professor Sun-Young Rha, Department of Medical Oncology at Yonsei Cancer Center, commented, “About 90% of metastatic gastric cancer patients are HER2-negative, and there has been a pressing need for new biomarker-targeted therapies. About 40% of HER2-negative patients are reported to be Claudin 18.2-positive, so the emergence of Vyloy, which selectively binds to Claudin 18.2, offers a new therapeutic possibility.”

- InterView
- "New landscape in DLBCL trt…Columvi·Polivy's bigger role"
- by Hwang, byoung woo Oct 10, 2025 06:06am
- Diffuse large B-cell lymphoma (DLBCL) has been known to lack new treatment options besides rituximab (MabThera) for over 20 years. Approximately one-third of DLBCL patients who undergo existing treatments (primarily R-CHOP) experience relapse or become non-responsive, and the second-line treatment has been limited to intensive chemotherapy and autologous stem cell transplant. However, the treatment paradigm is changing as innovative treatments, including antibody-drug conjugate (ADC), bi-specific antibodies, and CAR-T cell therapies, have emerged recently. During the meeting with DailyPharm, Professor Gareth Gregory of Monash University in Australia emphasized the role of treatments with new mechanisms, such as Polivy and Columvi, for older adults or patients who failed previous therapies. DLBCL still lacks treatments for older adults or transplant-ineligible patients…second-line treatment alternatives are needed Professor Gareth Gregory of Monash University in AustraliaAccording to Professor Gregory, the highest unmet needs for DLBCL are the second-line treatment. Exiting treatment data show that approximately one-third of all patients with DLBCL experience disease progression despite undergoing first-line treatments. To date, treatment options have been limited to intensive chemotherapy and autologous stem cell transplant. Professor Gregory said, "A significant proportion of DLBCL patients globally are elderly, aged 68 or older, and often have comorbidities, thus most of them are ineligible for autologous stem cell transplant," and added, "Given the high relapse rate characteristic of lymphoma, the response rate for patients receiving CAR-T or autologous stem cell transplant treatment in the second line is only about 21%, highlighting a significant need for more effective and safer therapeutic options." For this reason, interest has grown in treatments with new mechanisms, like bispecific antibodies. The need for monotherapy or combination therapy options, particularly for elderly patients or those who have failed previous treatments, is persistently being raised. Among the therapies currently driving a paradigm shift in DLBCL treatment are Polivy (polatuzumab vedotin) and the combination therapy of the bispecific antibody Columvi (glofitamab) with Gemcitabine and Oxaliplatin (GemOx). Particularly in the first-line setting, Polivy in combination with R-CHP is emerging as the new standard of care, replacing the traditional R-CHOP. For relapsed/refractory patients, the combination of the bispecific antibody Columvi and GemOx is gaining attention as a powerful alternative that shows long-term efficacy. Regarding this, Professor Gregory emphasized that the core of DLBCL treatment is not the 'effectiveness of a single agent,' but the 'design of the entire treatment journey.' For example, this involves a strategy in which Polivy reduces the risk of relapse in the first line, and Columvi increases the complete response rate in the second-line treatment setting, thereby extending treatment continuity. Professor Gregory said, "Polivy reduces the risk of relapse and death in first-line treatment, and Columvi offers the potential for long-term survival for relapsed patients," and added, "Both treatments are evolving to improve the patient's entire treatment journey." Polivy drives changes to the first-line setting...increased expectation of expanded reimbursement Polivy is evaluated as a key therapy for reducing the risk of recurrence associated with the conventional R-CHOPregimen and for improving long-term patient survival. Fortunately, it recently passed the Cancer Disease Review Committee (CDRC) in Korea, raising expectations for expanded reimbursement for first-line treatment. The 5-year follow-up data from the POLARIX study showed that the Polivy + R-CHP combination therapy improved both progression-free survival (PFS) and overall survival (OS) compared with conventional R-CHOP, and the rate of transition to subsequent treatments was significantly lower. Professor Gregory said, "Polivy combination therapy has been included in the first-line recommendations in guidelines in multiple countries, including Australia," and added, "Tolerability was similar to or better than R-CHOP, and we confirmed a long-term trend of reduced risk of relapse and death." Furthermore, Professor Gregory also pointed out that, given the characteristic of DLBCL, where the risk of relapse increases sharply if complete response is not achieved in the first line, the Polivy combination therapy is significant for its role in reducing treatment burden and medical resource consumption. He said, "Polivy combination therapy is significant not only for its treatment results but also for the efficiency of medical resources," and added, "Reducing relapse shortens the complex treatment process that leads to high-intensity therapies, and can also reduce patient hospitalization time, medical costs, and the loss of social productivity." Columvi offers long-term survival potential for relapsed patients...reimbursement remains a hurdle Meanwhile, in the second-line setting, Columvi is showing new possibilities. Columvi, a bispecific antibody, targets T cells and B cells simultaneously, achieving a high response rate even in relapsed/refractory patients with reduced chemotherapy response. In the 2-year follow-up analysis of the STARGLO Phase 3 trial, the Columvi + GemOx combination group had a 4-fold increase in PFS (13.8 months vs. 3.6 months) and a more than 2-fold improvement in complete response rate (58%) compared with the rituximab + GemOx group. Notably, 82% of patients who achieved remission maintained their response for at least 1 year, demonstrating the potential for long-term survival despite the fixed duration of therapy. Columvi's step-up dosing strategy is highlighted as a clinical feature that secures safety by minimizing immune-related side effects. Professor Gregory said, "Bispecific antibodies carry the risk of Cytokine Release Syndrome (CRS) due to immune system activation, but Columvi can be managed safely through step-up dosing and pre-treatment," and emphasized, "Columvi has the biggest advantage in that it is an 'off-the-shelf' therapy, meaning it can be administered immediately after diagnosis." He assessed, "Some patients in the STARGLO trial have maintained a long-term response for over four years, and for patients ineligible for CAR-T or transplant, the Columvi combination therapy offers a possibility of cure." Despite these therapeutic effects, the issue of utilization in Korea remains a reimbursement barrier. Polivy was stuck in a non-reimbursed state for five years after its 2020 approval, and Columvi also failed to pass the CDRC in December last year. Although Polivy initiated its first step toward reimbursement this year with discussions for first-line coverage, Columvi is still waiting for reimbursement listing for its second-line indication. Professor Gregory mentioned, "Effective treatment is meaningless if it is not delivered to the patient," and added, "Drugs with better accessibility and tolerability should realistically be reimbursed, especially compared to high-cost therapies like CAR-T, which are restricted to limited facilities." According to Professor Gregory, Australia has already approved reimbursement for the combination therapy of Columvi + GemOx for autologous stem cell transplant-ineligible patients, using the same criteria as those used in the clinical trial patient population. Finally, he said, "The ultimate goal of lymphoma treatment is to create patient-centered outcomes. If a treatment has sufficient accumulated clinical data, it should be applied to the field quickly," and added, "Institutional support is needed so that patients can receive the best care more safely and rapidly."

- Policy
- 'Low-price purchase incentive ineffective and detrimental'
- by Lee, Jeong-Hwan Oct 10, 2025 06:05am
- Rep. Joo-young Lee There is growing criticism that the government’s low-price drug purchase incentive policy should be overhauled due to its structural contradictions and low effectiveness. The low-price purchase incentive, which is linked to the market-based actual transaction price reduction system, focuses on price rather than quality. From the pharmaceutical companies’ perspective, the lower the actual transaction price (purchase price), the higher the likelihood of a price cut, rendering it difficult for them to actively participate in the policy. Some critics have also raised concerns that the system could be exploited for illegal drug rebate practices, calling for a review of whether it should be fundamentally reformed or even abolished. On the 3rd, Rep. Joo-young Lee of the Reform Party, who is also a member of the National Assembly’s Health and Welfare Committee, said, “The low-price purchase incentive system was designed to enhance the financial soundness of the national health insurance and reduce patient drug costs, but it has become an outdated policy that no one welcomes anymore.” Rep. Lee emphasized that if the government wants to establish a reasonable drug pricing system that both strengthens the pharmaceutical industry and ensures the stability of health insurance finances, it must first abolish policies that do not function in the actual healthcare field, such as low-price purchasing incentives. Under Article 22 of the Enforcement Decree of the National Health Insurance Act, the Health Insurance Review and Assessment Service (HIRA) currently operates the incentive system that pays medical institutions (hospitals, clinics, and pharmacies) 70% of the difference between the reimbursement ceiling price and the actual purchase price when they buy drugs of the same ingredient, dosage, and formulation at a lower price than the insurance ceiling. The program aims to reduce national health insurance expenditures, expand the use of generic drugs, curb excessive use of high-priced medications, and lower patient out-of-pocket costs. It has been in effect since 2010. However, Korean pharmaceutical companies and wholesalers argue that the system has failed to achieve these goals and contains inherent contradictions. They have consistently demanded major reforms or the abolition of the policy. Rep. Lee saw eye to eye on these concerns, urging the government to make substantive policy changes. At the industry level, critics argue that low-price purchasing incentives risk promoting an industry structure that prioritizes price over quality. This is because such incentives reward companies based on how cheaply they can procure drugs, rather than rewarding them for the quality of the drugs themselves. As long as the government maintains a policy that rewards cheaper supply, manufacturers will be incentivized to cut production costs and quality to produce low-cost drugs, sustaining a downward spiral in the market. The pharmaceutical industry and drug wholesalers argue that the low-price purchase incentive system inherently contains a contradiction, as it links incentives to reductions in actual transaction drug prices. They point out that the lower the actual transaction drug price becomes to qualify for the low-price purchase incentive, the greater the likelihood it will later be targeted for price reduction. Consequently, no one is willing to trade at lower drug prices. Healthcare institutions have long pointed out that for small and medium-sized hospitals, neighborhood clinics, and pharmacies—not large tertiary hospitals—the actual volume of low-price purchases is too small, resulting in a low perceived incentive effect. Critics note that over 80% of incentives are concentrated in large tertiary hospitals and mid-sized facilities, often benefiting institutions engaging in “one-won bidding” practices. Rep. Lee stated, “Both the low-price purchase incentive and the actual transaction price reduction systems are built on a price-based structure, not on generic drug quality. They contradict the government’s stated goal of fostering the pharmaceutical industry as a national growth engine and supporting global expansion.” Lee also warned of potential abuse of the system through illegal rebates or manipulation of prescription volumes to maintain sales of specific drugs. Rep. Lee concluded, “If the system has neither achieved its original goal of reducing health insurance expenditures nor contributed to the development of the pharmaceutical industry, the government should not leave it as is. Rather, the government should actively consider abolishing it. There is no reason to maintain a policy that no one supports and that only invites calls for reform or repeal.”
- Company
- Boehringer wins 'Jardiance' generic trademark dispute
- by Kim, Jin-Gu Oct 10, 2025 06:05am
- Product photo of Jardiance In a trademark dispute over the generic-name trademark for the SGLT-2 inhibitor diabetes treatment, 'Jardiance (empagliflozin),' the original manufacturer has won a ruling from the Intellectual Property Trial and Appeal Board (hereafter, the Board). Analysis suggests that the Board's rare decision to rule in favor of the original company was based on the high similarity between the English brand name, 'Jardiance,' and the disputed generic trademark, 'Jadiance.' According to the pharmaceutical industry on the 4th, the Intellectual Property Trial and Appeal Board recently issued a ruling of 'acceptance' in the trademark nullification trial filed by Boehringer Ingelheim against Shinil Pharma. The trademark in question is 'Jadiance,' which Shinil Pharma filed in March 2022. Shinil Pharma applied for this trademark with a focus on generics for both Jardiance and Jardiance Duo. The trademark was registered in August 2023. Shinil Pharma subsequently received a priority marketing authorization for a Jardiance Duo generic under the name 'Jadiance Duo.' However, just two months after the 'Jadiance' trademark was registered, Boehringer Ingelheim filed a nullity action in October 2023. The Board sided with the original manufacturer after approximately two years of deliberation. With generics expected to launch simultaneously after the expiration of the Jardiance substance patent on the 23rd of this month, Shinil Pharma is now unable to launch its generic product under the name 'Jadiance Duo' due to the loss of the trademark dispute. Shinil Pharma must either receive a reversal ruling through an appeal to the Patent Court or secure a new product approval under a different trademark to launch its generic. In this regard, Shinil Pharma holds trademarks such as 'Januglia,' 'Janumetia,' 'Januxr,' and 'Empagl,' which are presumed to be for Jardiance and Jardiance Duo generics, and these are not subject to the current dispute. The ruling is being called exceptional, given the Board's precedents. While global pharmaceutical companies have brought numerous legal actions to protect the trademarks of their original drugs, the Board and the Supreme Court have generally been lenient toward generic companies. More recently, in May, Novartis lost a trademark nullification trial it filed against Elyson Pharmaceutical, arguing that the generic name 'Entrelto' was too similar to its heart failure treatment, 'Entresto.' Similarly, Boehringer Ingelheim filed nullification trials in 2020 against Kwangdong Pharmaceutical's 'Dijenta' and Daewoong Pharmaceutical's 'Traceta' for being too similar to its diabetes treatment, 'Trajenta,' but those trials were also dismissed. Even the trademark dispute between 'Gliatirin' and 'Gliatamin' was ultimately decided in favor of the generic company by the Supreme Court. Meanwhile, the original company won the latest dispute, which is unlike past rulings and decisions. In this regard, the pharmaceutical industry notes that the Board found a high degree of similarity between Jardiance's English brand name, 'Jardiance,' and Shinil Pharm's 'Jadiance.' The global product name is 'Jardiance.' Both the U.S. FDA and the European Medicines Agency (EMA) approved the original empagliflozin-containing diabetes treatment under the product name 'Jardiance.' However, when Boehringer Ingelheim introduced the product in Korea, the company obtained product approval under the Korean spelling of "Jardiang" rather than the English pronunciation 'Jardiance.' Despite this, the approved English product name remains 'Jardiance,' identical to the global name. The company's registration with the Ministry of Intellectual Property only includes the Korean name "Jardiang", not 'Jardiance.' In this context, it is also explained that Boehringer Ingelheim only filed a trademark nullification trial against Shinil Pharma's 'Jadiance.' It has been confirmed that Shinil Pharma's 'Jadiance' is the only case currently involved in a trademark dispute with Boehringer Ingelheim over Jardiance and Jardiance Duo.
- Policy
- GLP-1 drug Ozempic passes reimbursement review
- by Jung, Heung-Jun Oct 10, 2025 06:05am
- Novo Nordisk’s GLP-1 receptor agonist Ozempic (semaglutide) has passed review by the Health Insurance Review and Assessment Service (HIRA)’s Drug Reimbursement Evaluation Committee, which acknowledged the drug as adequate for reimbursement. Ozempic contains the same active ingredient as the obesity drug Wegovy, but is indicated for diabetes. Meanwhile, the reimbursement scope for Janssen Korea’s prostate cancer drug Erleada (apalutamide) will be expanded. In addition to the existing indication of “metastatic hormone-sensitive prostate cancer (mHSPC),” the new coverage will include treatment for “high-risk non-metastatic castration-resistant prostate cancer (nmCRPC).” On October 2, HIRA held its 10th Drug Reimbursement Evaluation Committee meeting of 2025 to review applications for new drug reimbursement and expanded indications for drugs under risk-sharing agreements. Three drugs - Novo Nordisk’s Ozempic pre-filled pen (semaglutide 2 mg/1.5 mL, 4 mg/3 mL); Shinpoong Pharm’s Hyalflex Inj (hexamethylenediamine dihydrochloride bridged sodium hyaluronic acid gel) for knee osteoarthritis; and Mitsubishi Tanabe Pharma Korea’s Uplizna Inj (inebilizumab) for neuromyelitis optica spectrum disorder (NMOSD) – were reviewed. Ozempic was recognized as adequate for reimbursement “as an adjunct to diet and exercise for adults with type 2 diabetes inadequately controlled by existing therapies (in combination with other antidiabetic agents).” This marks the second time the drug has cleared reimbursement evaluation since 2023. While the first approval included a condition to accept a price below the assessed value, this latest decision carries no such condition. Novo Nordisk reportedly made substantial efforts by submitting supplementary data to HIRA for reimbursement and will now proceed to price negotiations with the National Health Insurance Service (NHIS). Uplizna was recognized as adequate for conditional reimbursement – allowed reimbursement when the company accepts a price below the assessed value—for treating “adult patients positive for anti-aquaporin-4 (AQP4) antibodies with neuromyelitis optica spectrum disorder.” The final listing will follow after the company completes price negotiations with the NHIS. Erleada’s reimbursement will expand to include “high-risk non-metastatic castration-resistant prostate cancer (nmCRPC).” The drug has been reimbursed since April 2023 for “metastatic hormone-sensitive prostate cancer (mHSPC).”

- Company
- Boryung acquires global rights to Taxotere for KRW 288B
- by Chon, Seung-Hyun Oct 02, 2025 06:13am
- Boryung acquired global rights to Sanofi's anticancer drug ‘Taxotere’ for up to KRW 287.8 billion. This investment, approaching KRW 300 billion, secures a revenue stream of approximately KRW 100 billion in annual sales. Boryung announced on the 30th that it has signed a global licensing agreement with Sanofi for the global business of the cytotoxic anticancer drug ‘Taxotere’ (docetaxel), including domestic and international rights, distribution rights, licensing rights, production rights, and trademark rights. The deal is valued at up to EUR 175 million (KRW 287.8 billion). Of this, EUR 161 million (KRW 264.8 billion) will be paid at closing, while the remaining EUR 14 million (KRW 23 billion) will be contingent on achieving certain contractual milestones. Through this acquisition, Boryung will take over the comprehensive business operations of Taxotere in 19 countries, including Korea, China, Germany, and Spain, as well as in Latin America and the Middle East, subject to approval by local regulatory authorities. Once regulatory procedures are completed, Boryung plans to manufacture Taxotere at its Yesan Campus and directly distribute and market the drug in global markets. Docetaxel is listed on the WHO Model List of Essential Medicines, and Taxotere is the original brand product of docetaxel. First approved by the U.S. FDA in 1995, Taxotere has been widely used for the treatment of various solid tumors, including breast cancer, prostate cancer, gastric cancer, and head & neck cancers. According to Sanofi, the product generated global sales of EUR 70 million (KRW 115.4 billion) last year. Even today, the clinical utility of Taxotere continues to expand, particularly in combination therapies, reinforcing its role as a key component in global cancer treatment. A Boryung representative explained, “Although the paradigm in oncology is shifting toward targeted and immuno-oncology therapies, cytotoxic agents remain a fundamental backbone of cancer treatment.” Boryung has previously taken over the domestic operations of global original oncology drugs such as Gemzar (2021) and Alimta (2023), successfully transitioning them to in-house production and ensuring stable supply. Boryung aims to advance as a global pharmaceutical company in the field of cytotoxic anticancer drugs through the acquisition of the Taxotere business. A Boryung official emphasized, “In the actual global market, repeated stockouts and supply disruptions of cytotoxic anticancer drugs are impacting patient treatment. Through this acquisition of the Taxotere global business, we plan to expand our differentiated portfolio in the cytotoxic anticancer drug field by stably establishing the global supply chain for these essential medicines, whose importance has grown.” Jeong-Gyun Kim, CEO of Boryung, said, “Boryung has gone beyond simple product acquisitions, internalizing manufacturing and formulation improvements to secure sustainable competitiveness. The Taxotere global business acquisition marks not only our third anticancer drug takeover after Gemzar and Alimta, but also the first time we have acquired global rights to an original medicine, paving the way for full-scale overseas expansion.” Kim added, “We will advance Taxotere's therapeutic value by expanding beyond simple technology transfer into comprehensive R&D, including follow-up formulation development, combination therapy strategies, and research into new indications. Through this, we aim to strengthen our differentiated portfolio in the cytotoxic oncology field, directly manufacture and distribute original anticancer medicines on the global stage, and reinforce our future growth engines.”.
- Company
- Severe asthma drug 'Fasenra' enters 'Big 5' gen hospitals
- by Eo, Yun-Ho Oct 02, 2025 06:11am
- Product photo of Fasenra 'Fasenra,' a treatment for severe asthma, is now available by prescription at tertiary general hospitals. According to industry sources, AstraZeneca Korea's Fasenra (benralizumab) passed the drug committees (DC) of 'Big 5' general hospitals, including Samsung Medical Center (Seoul), Seoul National University Hospital, Seoul Asan Medical Center, Seoul St. Mary's Hospital, and Sinchon Severance Hospital. Fasenra was included in the insurance reimbursement list in July of last year. The drug can be reimbursed when treating patients with severe eosinophilic asthma who are inadequately controlled despite treatment with high-dose inhaled corticosteroid-long-acting beta-agonist (ICS/LABA) and long-acting muscarinic antagonist (LAMA). Specifically, the following criteria should be met: ▲Within the year before starting treatment, the eosinophil count in the blood was 300 cells/㎕ or higher, and within the first year of treatment, systemic oral corticosteroids (OCS) were required for acute exacerbations four or more times, or within 6 months before starting therapy, systemic oral corticosteroids were continuously administered, or ▲The eosinophil count in the blood was 400 cells/㎕ or higher within the year before starting treatment. Systemic corticosteroids were required for acute exacerbations three or more times within the first year of treatment. Severe eosinophilic asthma accounts for approximately 84% of severe asthma cases. It involves frequent exacerbations and may lead to reduced quality of life despite treatment with high-dose inhaled corticosteroids and other conventional therapies. In particular, when symptoms are not controlled, even with asthma controllers, oral steroids may be necessary. However, long-term use of these medications is associated with systemic side effects such as osteoporosis, hypertension, and diabetes. Therefore, biological agents are recommended to reduce the dosage of these treatments. Fasenra is a targeted biologic agent that binds directly to interleukin-5 receptor alpha (IL-5Rα) expressed on eosinophils' surface, inducing cell apoptosis. It has been demonstrated to reduce blood eosinophil counts rapidly within one day of administration. Meanwhile, in the results from the global Phase 3 SIROCCO clinical trial, enrolling 1,205 severe eosinophilic asthma patients worldwide, including those in Korea, Fasenra administered at 8-week intervals showed a 51% reduction in annual asthma exacerbation rates compared to placebo after 48 weeks of treatment. In the CALIMA study, Fasenra treatment also resulted in a 28% reduction in annual asthma exacerbation rates compared to placebo.
- Company
- CKD to exclusively sell Bayer’s heart failure drug Verquvo
- by Chon, Seung-Hyun Oct 02, 2025 06:11am
- Chong Kun Dang announced on the 1st that it has signed an exclusive sales agreement with Bayer Korea for the chronic heart failure treatment ‘Verquvo’. Under the agreement, Chong Kun Dang will handle the exclusive distribution, sales, and marketing of Verquvo at domestic hospitals and clinics starting this month. Verquvo is a treatment for symptomatic chronic heart failure in patients with a left ventricular ejection fraction (LVEF) of less than 45%. It is the world’s first soluble guanylate cyclase (sGC) stimulator approved for chronic heart failure. The drug directly stimulates the nitric oxide–sGC–cGMP pathway, demonstrating efficacy in improving vascular function and enhancing cardiac structure and function. In global clinical trials, Verquvo was shown to reduce the risk of cardiovascular death and hospitalization due to heart failure in high-risk chronic heart failure patients who had experienced worsening of symptoms despite standard therapy. It is evaluated as presenting a new treatment paradigm for high-risk patient groups, acting through a mechanism distinct from existing neurohormone blockade-centered therapies. Chong Kun Dang and Bayer Korea have been long-standing partners. Since 2005, they have co-promoted antibiotics such as Cifrobay and Avelox, and since last year, they have co-marketed Kerendia, a treatment for chronic kidney disease associated with type 2 diabetes. In February 2024, Chong Kun Dang also began exclusive sales of Bayer’s progressive hepatocellular carcinoma treatments Nexavar and Stivarga in Korea, further strengthening their partnership. Young-Joo Kim, President of Chong Kun Dang Pharma, said, “Chong Kun Dang has been leading the cardiovascular disease treatment market based on its extensive experience and expertise in the field. With the exclusive sales of Verquvo, we will be able to establish a differentiated portfolio in the field of chronic heart failure treatment and provide patients with broader therapeutic options.” JinA Lee, President/CEO of Bayer Korea, said, “Since Verquvo was reimbursed under the national health insurance in September 2023, it has become a cornerstone treatment for patients experiencing worsening heart failure symptoms. Through our collaboration with Chong Kun Dang, we hope more Korean patients with chronic heart failure can benefit from Verquvo’s proven clinical value.”