- LOGIN
- MemberShip
- 2026-04-28 20:54:54

- Policy
- MOHW proposes an alternative to law on generic substitution
- by Lee, Jeong-Hwan Aug 19, 2025 06:11am
- A revision to the Pharmaceutical Affairs Act that would allow pharmacies to report generic substitution to the Health Insurance Review and Assessment Service (HIRA) through its work portal is likely to pass the National Assembly. The background to the sudden increase in the likelihood of legislation is that the Ministry of Health and Welfare submitted its own alternative proposal to the National Assembly regarding the bill to simplify generic substitution by pharmacies and changed its position to conditional approval (acceptance) on the premise that its alternative proposal would be adopted. If the National Assembly accepts the alternative proposal submitted by the MOHW, the simplification of post-notification of generic substitution will be legislated in the Pharmaceutical Affairs Act, a law, rather than as an Enforcement Rule, a subordinate law. The alternative submitted by the MOHW stipulates that the Minister of Health and Welfare shall be able to support related administrative tasks, such as post-notification of generic substitution, and that such support tasks may be entrusted to the Health Insurance Review and Assessment Service. Rather than specifying the method of post-notification of generic substitution in the law, the proposal broadly stipulates that the MOHW shall provide support and legalize the framework for entrusting the MOHW's duties to HIRA. On the other hand, HIRA maintained its cautious stance, citing concerns for patient safety in the event of an accident involving a medication misadventure and the lack of legal grounds for entrusting the task to HIRA. This follows the Ministry of Health and Welfare’s submission, on the 18th, of its opinion on the amendment to the Pharmaceutical Affairs Act (aimed at simplifying generic substitution) to the National Assembly Health and Welfare Committee’s First Legislative Subcommittee. The law to simplify generic substitution was proposed by Representative Young-seok Seo of the Democratic Party of Korea and Representatives Sujin Lee and Byung-deok Min of the same party. The Health and Welfare Committee plans to hold the 1st Subcommittee meeting on the 19th to review the bill. The main contents are a provision to change the term “substitute dispensing” to “generic name substitutions” and a provision to expand the scope of post-notification of substitute prescriptions to HIRA. The change in terminology provision has been strongly opposed by the medical community, including the MOHW, and it is unlikely to pass the National Assembly. The provision to simplify generic substitution by expanding the scope of post-notification was once rejected by the subcommittee after the MOHW expressed its need for cautious review. However, the MOHW submitted a legal alternative it had drafted itself to the subcommittee and changed its position to support the legislation on the condition that it be passed by the National Assembly as is. The simplification of post-notification of generic substitution is expected to take effect on February 2 next year, as MOHW has already confirmed the revision of the Enforcement Rules of the Pharmaceutical Affairs Act, and the possibility of the National Assembly revising the Pharmaceutical Affairs Act has also increased. The MOHW has once again expressed its cautious stance on legislation that would require HIRA to notify prescribing doctors of the results of post-notification of generic substitution by pharmacies. The MOHW argued that this is not in line with the purpose and scope of HIRA's establishment and that the increase in the notification period could raise concerns about the safety of drug use. The MOHW also pointed out that the amendment to the Enforcement Rules of the Pharmaceutical Affairs Act allows post-notification via HIRA's information system in addition to telephone and fax. However, in consideration of the need to clarify the legal basis for the HIRA information system currently being developed by the MOHW and the necessity of substitute dispensing in response to drug supply uncertainties, the MOHW stated that an alternative plan would be necessary to establish a basis for HIRA's policy support. So the alternative submitted by the MOHW to the National Assembly stipulates the establishment of “support for generic substitution under Article 27-2 of the Pharmaceutical Affairs Act” and, in Paragraph 1, stipulates that the Minister of Health and Welfare may provide support for matters specified by MOHW ordinances, such as support for post-notification of generic substitutions. Paragraph 2 allows the Minister of Health and Welfare to entrust HIRA with the task of providing support for post-notification of generic substitution, and paragraph 3 stipulates that the details and methods of support and other necessary matters shall be determined by MOHW ordinances.. However, HIRA maintained its cautious stance on the bill. The logic behind its need for cautious review is that if post-notification generic substitution is carried out through HIRA, the notification period will increase, and if an accident occurs due to a lack of awareness on the part of doctors, this will raise concerns about patient safety. In addition, HIRA argued that it can only perform tasks entrusted to it by other laws and regulations in accordance with Article 63 of the National Health Insurance Act, and therefore, a separate basis for entrusting tasks related to generic substitution notifications is necessary. Furthermore, it also argued that unique identification numbers in accordance with the Personal Information Protection Act, regulations on the handling of sensitive information, and provisions exempting HIRA from liability for medication misadventure are necessary. d The Korean Pharmaceutical Association in favor, Korean Medical Association and Korean Hospital Association oppose The Korean Pharmaceutical Association supported the bill. On the other hand, the Korean Medical Association and the Korean Hospital Association maintained their opposition. The Korean Pharmaceutical Association predicted that allowing HIRA to notify doctors and dentists electronically would eliminate administrative inconveniences and streamline procedures, thereby promoting generic dispensing, considering the difficulty of notifying doctors and dentists when pharmacists substitute drugs due to factors such as failure to include fax numbers or failed phone connections. The Korean Medical Association argued that pharmacists could arbitrarily change medications without the consent of doctors, which could undermine the right of doctors to prescribe medications, make it difficult to respond quickly to adverse drug reactions, and raise questions about the accountability of the dispensed generic substitutions and their side effects. The Korean Hospital Association also opposed the bill, arguing that notifying just the HIRA of generic substitutions would make it difficult for doctors and pharmacists to share information about generic substitutions, which could delay the necessary treatment for patients in cases of inappropriate generic substitutions.
- Opinion
- [Reporter's View] Clarify support for unstable supply drugs
- by Lee, Hye-Kyung Aug 19, 2025 06:11am
- The Ministry of Food and Drug Safety is reportedly preparing support measures to stabilize the supply of national essential medicines and other items with unstable supply. The MFDS initially announced its intention to specify the urgent measures required by the industry to stabilize supply, such as emergency imports, custom manufacturing, and administrative support, within the first half of this year. However, it was not specified within the first half of the year, and it is said that the authorities are still considering how to specify and disclose the measures. Support for stabilizing the supply of drugs is provided by the Pharmacuetical Management Support Team of the MFDS. The team was newly established in March 2024 after the tasks previously handled by the Pharmaceutical Policy Division within the Pharmaceutical Safety Bureau were transferred due to the growing need to respond to public health crises caused by COVID-19. The Pharmaceutical Management Support Team will operate on a temporary basis until January 31, 2027, and is currently responsible for securing a stable supply system for essential medicines, ensuring the swift and stable supply of medical products in crisis situations, as well as other related tasks. In particular, it is responsible for tasks such as the designation of essential medicines, reporting on supply disruptions or shortages, monitoring supply and demand, providing administrative support and communicating with companies, and taking measures to maintain the supply of discontinued items. In this process, the pharmaceutical industry has consistently requested that administrative support measures be clearly stipulated in writing. Support measures are necessary to ensure the continued supply of essential medicines in cases of supply disruptions, as there have been instances where price increases were implemented for certain items. Therefore, there is a growing demand for the sharing of individual cases to clarify what support can be provided for which specific items. The MFDS is also preparing guidelines that include various administrative support measures, but the problem is that it is struggling with whether to use these guidelines internally or disclose them externally. If the guidelines are disclosed externally, administrative support may be applied to all items, which would inevitably increase the administrative burden. In the case of stabilizing the supply of pharmaceuticals, support measures may vary by product, and if the guidelines are made public, some companies may raise issues regarding the varying standards. However, formalizing administrative support could be one of the most effective measures to help companies stabilize supply. Companies may need to bear the burden of administrative costs and continue producing items that could otherwise be discontinued, so it is crucial for the government to clarify the specific support it can provide. Therefore, if guidelines are established, they should be publicly disclosed rather than kept internal, and administrative support measures should be properly established through a case-by-case approach.
- Company
- Will Wegovy patients switch to Mounjaro? Doctors skeptical
- by Moon, sung-ho Aug 19, 2025 06:11am
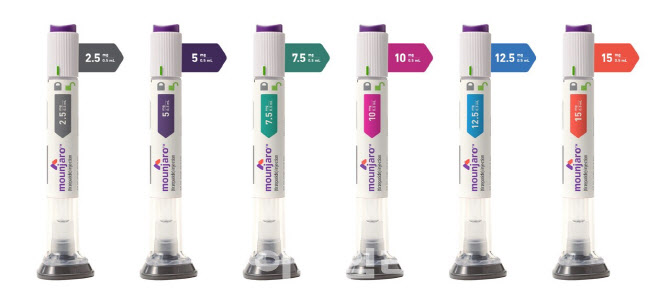
- With the anticipated launch of Lilly Korea's diabetes and obesity treatment Mounjaro (tirzepatide) in mid-August, its full-scale competition with Novo Nordisk's Wegovy (semaglutide) is expected to unfold. Amid the imminent competition, the issue of whether to switch between the two drugs has emerged as a key point of contention in clinical settings. #According to industry sources on the 11th, Lilly Korea plans to launch Mounjaro Pre-filled Pen (tirzepatide, hereinafter referred to as Mounjaro) 2.5 and 5 mg for patients with type 2 diabetes and obesity in Korea in mid-August. Mounjaro is currently used in Korea as an adjunct to diet and exercise (as monotherapy or combination therapy) to improve blood glucose control in adult patients with type 2 diabetes, as well as in adult patients with obesity (initial BMI ≥ 30 kg/m2) or one weight-related comorbidity (hypertension, dyslipidemia, type 2 diabetes, obstructive sleep apnea, or cardiovascular disease) for chronic weight management as an adjunct to a low-calorie diet and exercise therapy. For both indications, the recommended starting dose is 2.5 mg once weekly (for treatment initiation, not for blood glucose control or weight management), followed by 5 mg once weekly starting at week 4. If additional dose adjustment is required, increase by 2.5 mg after at least 4 weeks of treatment at the current dose, with a maximum dose of 15 mg once weekly. The attention is now focused on the competition between Wegovy and Mounjaro in clinical practice. A key point of interest in this process is whether patients previously treated with Wegovy will switch to Mounjaro. Although there are no direct comparative studies, based solely on the results revealed during the development process, the weight loss effect of Mounjaro appears to be more pronounced. Considering this, some patients who were previously receiving Wegovy may express a willingness to switch to Mounjaro. In response, Lilly Korea stated that the decision to change the type of medication or treatment method prescribed to patients should be made through discussion between healthcare professionals and patients. A representative from Lilly Korea stated, “We have not conducted any studies on switching from GLP-1 RA to Mounjaro for chronic weight management on our own. However, we have a Phase IV clinical trial called SURPASS-SWITCH-2, which involved switching from GLP-1 RA (semaglutide, dulaglutide, or liraglutide) to Mounjaro in adult patients with type 2 diabetes for 12 weeks. The study was not designed to provide long-term results, efficacy comparisons, or optimal switching strategies, as these may vary depending on individual characteristics." In clinical practice, not many patients are expected to switch from Wegovy to Mounjaro, considering how the initial dose of Mounjaro is 2.5 mg. Chul-Jin Lee, President of The Korean Society For the Study of Obesity (Joeun Family Clinic), said, “explained, “Just because Mounjaro has been launched does not mean that patients who were previously receiving Wegovy will easily switch treatments in the short term. This is because patients who were receiving high doses of Wegovy may experience relatively lower efficacy when switching to 2.5 mg of Mounjaro.” Lee said, “If Mounjaro is released, it should be offered first to new patients. The 2.5 mg dose has an advantage in terms of price for patients as it is not covered by insurance, but for the 5 mg dose and above, the volatility of the range of the non-reimbursed price must also be considered.” Meanwhile, the supply price for Mounjaro has recently been finalized. The currently known domestic supply prices for Mounjaro are KRW 278,066 for 2.5mg, KRW 369,307 for 5mg, and KRW 521,377 for both 7.5mg and 10mg. The dosage forms to be launched in Korea this month are 2.5mg and 5mg. For reference, when Novo Nordisk launched Wegovy in Korea last year, the supply price was set at KRW 372,025 per month regardless of dosage. Accordingly, patients who receive Wegovy as a non-reimbursable drug, mainly at outpatient clinics, must pay an average of KRW 500,000 to 600,000 per month. Considering this, when the supply price is taken into account, the non-reimbursed price of 2.5 mg of Mounjaro is expected to be cheaper than Wegovy, but 5 mg is expected to be priced similar to the monthly cost of Wegovy.
- Company
- PARP inhibitor 'Lynparza' reattempts prostate cancer reimb
- by Eo, Yun-Ho Aug 18, 2025 06:03am
- Product photo of LynparzaAttention is focused on whether there will be progress in the expanded reimbursement of the PARP inhibitor 'Lynparza' for prostate cancer. According to industry sources, AstraZeneca Korea's PARP (Poly ADP-ribose polymerase) inhibitor Lynparza (olaparib) is expected to be considered for the Health Insurance Review & Assessment Service (HIRA)'s Cancer Disease Review Committee next month (September). The indications for review are two: ▲Treatment of adult patients with BRCA-mutated metastatic castration-resistant prostate cancer (mCRPC) who have experienced disease progression after treatment with a novel hormonal agent ▲Combination therapy with abiraterone and prednisolone in adult patients with newly diagnosed mCRPC who have not received prior chemotherapy. This is Lynparza's second attempt to get reimbursement for its prostate cancer indication. AstraZeneca had previously tried in 2022 to get reimbursement for BRCA-mutated prostate cancer, but the application was withdrawn after failing to reach an agreement during the post-Cancer Disease Review Committee procedure. Therefore, it remains to be seen if this second round of reimbursement discussions will be successful. Meanwhile, the efficacy of Lynparza in prostate cancer has been proven through two Phase 3 trials: the PROfound study and the PROpel study. A subgroup analysis of the PROfound study in mCRPC patients with BRCA1/2 mutations showed that in patients who had progressed after prior abiraterone or Xtandi (enzalutamide) treatment, Lynparza significantly improved radiographic progression-free survival (rPFS) to a median of 9.8 months, compared to 3.0 months for the control group. Overall survival (OS) was also improved, with a median of 20.1 months versus 14.4 months for the control group. The PROpel study evaluated the combination therapy of Lynparza and abiraterone in mCRPC patients who had not received prior chemotherapy. The study showed that the combination therapy reduced disease progression or the risk of death by 34% compared to abiraterone monotherapy. The median rPFS was extended by 8.2 months in the combination group, reaching 24.8 months compared to 16.6 months in the abiraterone monotherapy group.

- Company
- Four CAR-T therapies to compete for 'Kymriah's mkt position'
- by Whang, byung-woo Aug 18, 2025 06:02am
- With the third new CAR-T therapy, 'Yescarta,' obtaining marketing authorization in Korea, market competition for treating relapsed or refractory Diffuse Large B-cell Lymphoma (DLBCL) is anticipated. With Kymriah (tisagenlecleucel) from Novartis Korea being the only option currently reimbursed, the impact of Yescarta, which has shown prominence in the global CAR-T market, is highly anticipated. Furthermore, with the expected approval of Curocell's next-generation CAR-T therapy, Rimqarto, this year, various factors are expected to be considered in the choice of DLBCL treatments. Product photo of YescartaThe Ministry of Food and Drug Safety (MFDS) announced the marketing authorization for Gilead Sciences Korea's Yescarta (axicabtagene ciloleucel) on the 13th. Yescarta received authorization for the treatment of 'adult patients with Diffuse Large B-cell Lymphoma (DLBCL) who relapse or are refractory within 12 months of first-line chemoimmunotherapy' and 'adult patients with relapsed or refractory DLBCL and Primary Mediastinal B-cell Lymphoma (PMBCL) after two or more lines of systemic therapy.' Administration is required to be performed only at certified medical institutions, and certified medical institutions must have tocilizumab readily available on-site. The authorization also specifies that at least two doses of tocilizumab per patient must be secured before Yescarta administration in case treatment for Cytokine Release Syndrome (CRS) is needed. Yescarta is currently a leading therapy in the CAR-T treatment market. This is because Yescarta has secured a broader range of indications than Kymriah, which is limited to third-line therapy. First approved by the U.S. FDA as a third-line therapy in October 2017 and by the EU in 2018, Yescarta has since expanded its scope to include second-line therapy. In 2021, it became available to treat follicular lymphoma. In terms of sales, its revenue surged by 67%, from $695 million (approximately KRW 966.4 billion) in 2021 to $1.16 billion (approximately KRW 1.6127 trillion) in 2022. The growth continued, reaching $1.498 billion (approximately KRW 2.0827 trillion) in 2023, a 29% increase from the previous year. However, since the rapid growth of late entrants like Carvykti (ciltacabtagene autoleucel, Janssen) and Breyanzi (lisocabtagene maraleucel, BMS) starting last year, the pace of revenue increase has slowed. In its recently announced Q2 sales for this year, Gilead reported that Yescarta recorded $393 million (approximately KRW 550 billion), a 5% decrease from the previous year. The reason for the anticipation surrounding Yescarta is its indication. Kymriah's indication is for 'adult patients with relapsed or refractory DLBCL after two or more lines of systemic therapy.' For this reason, Gilead Sciences Korea is reportedly planning to prepare for reimbursement for the newly approved second-line therapy, rather than the third-line therapy, as Kymriah is already reimbursed for the third-line setting, making reimbursement for Yescarta at this stage less difficult. While new drugs of bispecific antibodies are receiving approval and reimbursement, CAR-T therapies still hold a preemptive treatment status. If Yescarta receives reimbursement, it is expected to quickly establish itself in the domestic market, given its global influence. Curocell Prepares for Rapid Commercialization…"Efficacy and Supply Still the Advantages" Another variable is that Curocell's Rimqarto is awaiting approval. Rimqarto has been selected as the second drug for the Ministry of Health and Welfare's 'Concurrent Application-Evaluation-Negotiation Pilot Program' and is currently undergoing review by the MFDS. While there are observations that the approval might be delayed beyond the initially expected third quarter due to delays in the review process, as it is the first domestically developed next-generation CAR-T therapy in Korea, the prevailing view is that approval will likely happen within the year. Once approved, it is expected to have an advantage in reimbursement entry because the reimbursement evaluation and price negotiation will be conducted simultaneously with the MFDS approval application process. In particular, the company is preparing for rapid commercialization by establishing a comprehensive solution, 'CUROLINK,' which tracks and manages the entire process from prescription to administration in real time. However, Rimqarto must keep Yescarta's approval in check, as it has the same indication as Kymriah. Concerning this, Curocell's position is that its competitiveness will remain valid because, when compared to Curocell's Rimqarto, Yescarta, and Kymriah are still inferior in terms of supply period, efficacy, and safety in the domestic market. A Curocell official stated, "Yescarta's entry into the Korean market and the emergence of other competing products like bispecific antibodies are always open possibilities," and added, "Yescarta still requires sending the patient's blood overseas for CAR-T manufacturing and then re-importing it into Korea, placing it in an inferior position compared to Curocell's Rimqarto in terms of rapid supply." Furthermore, according to an IR document released by Curocell, Yescarta showed a complete remission in 54% of patients who participated in the clinical trial. In contrast, Rimqarto's clinical trial showed a complete remission in 67% of participants. The Curocell official explained, "Yescarta's CR rate of 54% was far superior to Kymriah's 40%, which led to a decrease in Kymriah's sales and an increase in Yescarta's sales. However, when Breyanzi, with a CR rate of 53%, entered the market with a similar CR rate to Yescarta, it began to share the market, causing a slight decrease in Yescarta's sales." And added, "Considering this, Rimqarto's CR rate of 67% is higher than both Yescarta and Breyanzi. Given that this is a one-time reimbursed treatment for late-stage cancer patients, We believe Rimqarto will have a competitive advantage in the market."
- Company
- New Praluent dosage form available at general hospitals
- by Eo, Yun-Ho Aug 18, 2025 06:02am
- A new dosage form of the PCSK9 inhibitor ‘Praluent’ may be prescribed at general hospitals in Korea. According to industry sources, a pen formulation of Sanofi-Aventis Korea's primary hypercholesterolemia and mixed dyslipidaemia treatment Praluent (alirocumab), Praluent Pen 300mg inj, has been approved by the Drug Committees (DCs) of tertiary hospitals in Korea, including Seoul National University Hospital, Seoul Asan Hospital, and Sinchon Severance Hospital. Praluent Pen Inj 300mg maintains similar LDL-C lowering effects to the existing dosages while enabling administration at four-week (Q4W) intervals, thereby improving treatment convenience and patient compliance. This drug is the only PCSK9 inhibitor in Korea available in three doses (75 mg, 150 mg, and 300 mg), and all doses are indicated for reducing cardiovascular risk in patients with atherosclerotic cardiovascular disease (ASCVD). PK/PD studies confirmed the LDL-C-lowering efficacy of Praluent Pen 300mg, with LDL-C reduction effects observed as early as day 3 after a single dose of Praluent Pen 300mg injection, reaching a maximum average reduction of 73.7% by day 22. This LDL-C-lowering effect was maintained for up to 43 days, demonstrating a longer duration of effect compared to the existing Praluent 75mg Pen (8 days) and 150mg (15 days). Praluent 300 mg has been listed for reimbursement in Korea since April. As a result, Praluent Pen Inj may be used with reimbursement when administered in addition in ▲patients with primary hypercholesterolemia and mixed dyslipidemia who have received insufficient response to combination therapy with statins and ezetimibe (LDL-C levels not reduced by at least 50% from baseline or LDL-C ≥ 100 mg/dL) and statin intolerance ▲Patients with atherosclerotic cardiovascular disease who have received maximum tolerated doses of statins and ezetimibe but have had an inadequate response (LDL-C levels not reduced by at least 50% from baseline or LDL-C ≥ 70 mg/dL). In a Phase III trial (CHOICE 1) involving patients who were taking the maximum tolerated dose of statins, patients who were taking the maximum tolerated dose of statins in combination with other lipid-lowering agents, and patients who were not taking statins, the starting does of Praluent Pen 300 mg Inj demonstrated efficacy, long-term safety profile, and tolerability. According to the study, at week 24, the LDL-C reduction effect of Praluent 300 mg Inj plus statin therapy compared to placebo was 58.8%, showing a statistically significant result compared to the placebo group (0.1% reduction). In the case of its indication for atherosclerotic cardiovascular disease, the drug demonstrated efficacy iin the ODYSSEY OUTCOMES study. The study included 18,924 adult patients with acute coronary syndrome (ACS), including myocardial infarction and unstable angina. The results showed that Praluent significantly reduced the risk of major adverse cardiovascular events (MACE) by 15% compared to the placebo group. A trend toward a reduction in all-cause mortality was also observed as a secondary endpoint, which was nominally significant in hierarchical statistical testing. In addition, y, LDL-C achieved its maximum reduction effect at week 4 of Praluent treatment and maintained an average reduction of 54.7% after 4 years of treatment. LDL-C reduction was observed in 89% of patients receiving high-intensity statins in combination with Praluent.

- Policy
- Periodic drug price system reform included as national task
- by Lee, Jeong-Hwan Aug 18, 2025 06:02am
- President Lee Jae-myung attended the National Planning Committee The government has selected a policy to improve the drug price calculation system and establish a periodic drug price adjustment mechanism as part of its national policy task, with the goal of ensuring the sustainability of health insurance, and is pushing ahead with its implementation. Depending on the specific details of the national policy task to be established in the future, it is noteworthy that this could lead to additional post-marketing management regulations, such as a price reduction mechanism for generic drugs with expired patents, which is a highly concentrated area in the domestic pharmaceutical industry. To build a primary care-based health and care system, the government plans to gradually expand the local primary care physician model starting next year (2026) and adopt the institutionalization of telemedicine and public electronic prescription transmission systems as national policy tasks through social consensus. According to the National Assembly and the medical community on the 13th, the National Planning Committee has adopted such policy directions as national policy tasks. Transition to a sustainable healthcare system The National Planning Committee proposed improving the drug price calculation system and establishing a periodic drug price adjustment mechanism as a means of stabilizing the sustainability of the national health insurance budget, in order to curb expenditure factors that worsen the healthcare system, such as budget leakage. The government has been managing national health insurance drug expenses by adjusting the insurance ceiling price (drug price) of new and generic drugs through the post-marketing drug management system. In particular, following research conducted last year by Professor Dong-sook Kim’s research team at Kongju National University under the Yoon administration, the Ministry of Health and Welfare recently commissioned and launched additional research on the generic drug price system following the election of President Jae-myung Lee and the appointment of MOHW Minister Eun-kyung Jeong. The ministry has entered into a direct contract with a research team led by Professor Seon-mi Jang of the College of Pharmacy at Gachon University to conduct a study titled “Reform of the Drug Price Model (Focusing on Generic Drugs).” The results of this study is expected to serve as a basis for the government's drug price reductions. The Ministry of Health and Welfare under the Lee administration also agrees that it is necessary to establish financial leeway for national health insurance by adjusting the prices of old drugs with uncertain efficacy, so there is great social interest in whether new drug price reduction mechanisms, such as the overseas reference pricing reevaluation measures will emerge. However, there are also opinions that it is difficult to judge whether generic drug prices in South Korea are high or low compared to other developed countries. The Korean pharmaceutical industry is dissatisfied that the government is focused solely on establishing policies to reduce drug prices and is relatively neglecting policies that award premiums on drug prices. Ultimately, the pharmaceutical industry is expected to respond depending on how the details and direction of the national policy agenda are decided. Expansion of telemedicine and establishment of a public electronic prescription system The National Planning Committee has also selected the promotion of public health through primary care-based health management as a national policy task. The goal is to extend the healthy life expectancy of citizens by improving the management rate of chronic diseases through measures such as the expansion of local primary care physicians, the expansion of telemedicine, and the establishment of a public electronic prescription transmission system. The community-based local primary care physician model is planned to be expanded in phases starting in 2026, with the core elements being the provision of comprehensive health management based on multidisciplinary teams and innovative compensation based on performance. The Democratic Party of Korea has submitted a bill to the National Assembly to strengthen primary care and institutionalize the primary care physician system, titled the “Special Act on Strengthening Primary Care” (“Primary Care Special Act”, proposed by Representative In-soon Nam), ahead of the implementation of the Integrated Care Act. The Integrated Care Act and the Primary Care Special Act are expected to serve as the legal and administrative basis for building a society that provides national health care based on primary care. In particular, the National Planning Committee will institutionalize telemedicine based on social consensus and establish a system of telemedicine and remote consultation at public health centers targeting medically underserved areas in rural and fishing villages. Four bills to formally institutionalize telemedicine pilot projects (by Reps. Bo-yoon Choi, Jae-joon Woo, Jin-sook Jeon, and Chil-seung Kwon) are pending in the National Assembly, and the Health and Welfare Committee is expected to review them in the near future. The first subcommittee review is expected to take place as early as the 19th. The National Planning Committee is also expected to include a plan to establish and operate a public electronic prescription transmission system in its national policy task from 2027, based on opinions gathered from the medical community and the Pharmaceutical Association, in line with the institutionalization of telemedicine. The institutionalization of telemedicine and the introduction of public electronic prescriptions were also part of President Jae-myung Lee 's presidential campaign promise. Meanwhile, the National Planning Committee's national policy task must undergo final review by the relevant government ministries and the State Council before it can be finalized.
- Company
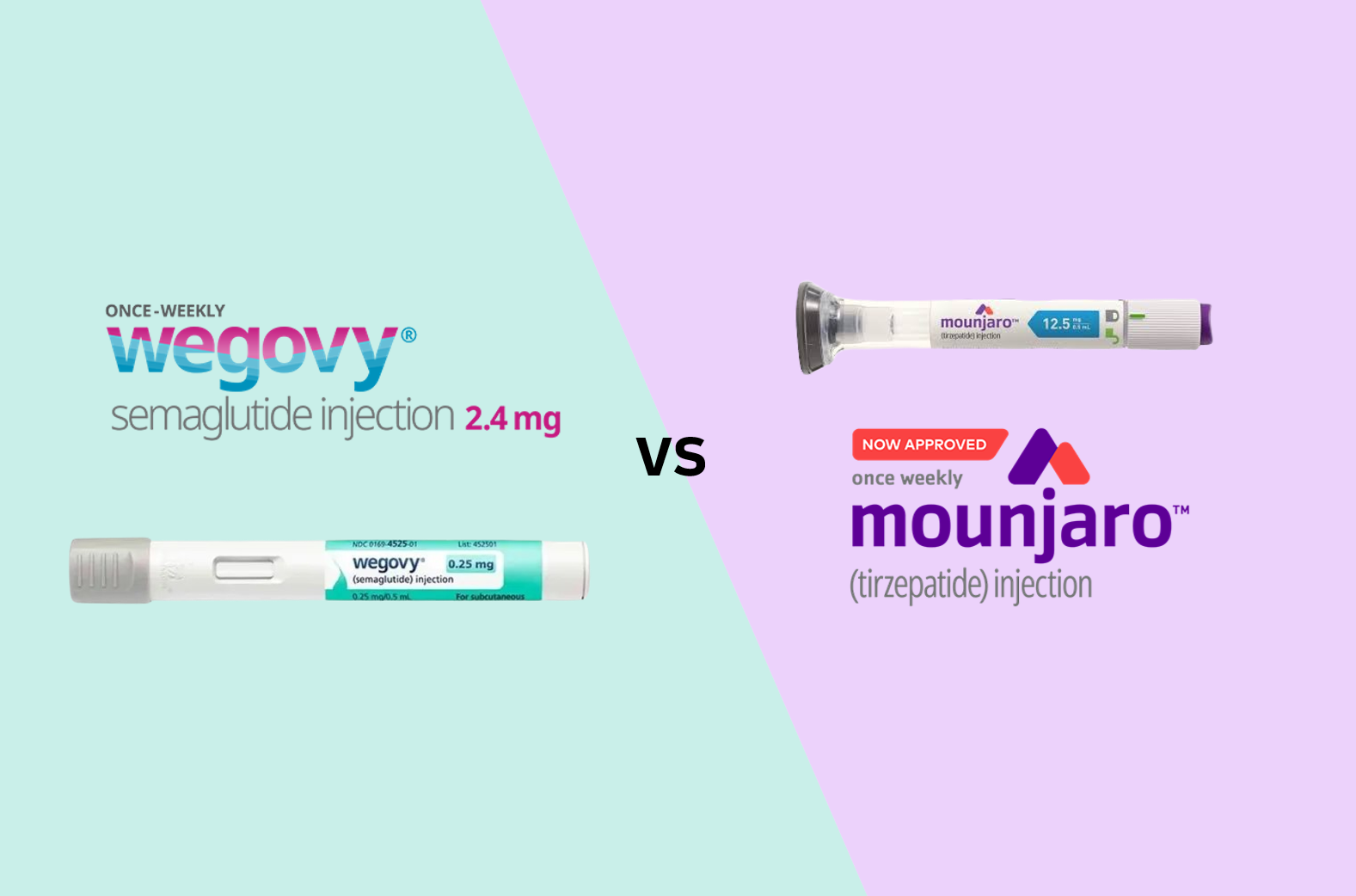
- Will Korean companies join in on the sales of obesity drugs?
- by Kim, Jin-Gu Aug 18, 2025 06:02am
- With the domestic launch of the GLP-1 class obesity treatment Mounjaro (tirzepatide) near, its sales and marketing competition with Wegovy (semaglutide) is already heating up. Lilly has completed preparations for the launch by hiring over 30 additional medical and marketing representatives to handle sales of Mounjaro. Additionally, the possibility of joint sales with domestic pharmaceutical companies is being raised. Novo Nordisk has countered by reducing the domestic supply price of Wegovy by up to 40%. Their discussions on joint sales with Chon Kun Dang are also reportedly ongoing. Mounjaro to be released next week, adds 30 sales staff... Large-scale launch symposium announced According to industry sources on the 14th, Lilly's Mounjaro will be distributed in Korea starting next week. Lilly recently sent a notice to major domestic distributors stating that its supply will begin on the 18th. Hospitals and clinics are expected to receive the product two days later on the 20th. Given the product's record-breaking sales abroad, Lilly Korea is also highly anticipating its launch. In preparation for the launch of Mounjaro, Lilly has hired over 30 additional employees dedicated to sales and marketing for the product. A large-scale launch symposium is also planned. Lilly intends to hold an in-person symposium on the 25th and 26th, followed by an online symposium on the 27th. The company has gone to great lengths to invite Bruno Halpern, the next president of the World Obesity Federation and chief director of the Obesity Centre of Hospital 9 de Julho, São Paulo in Brazil, as a keynote speaker. For now, Lilly Korea plans to directly sell Mounjaro, but the possibility of joint sales with domestic pharmaceutical companies remains open. In the pharmaceutical industry, one major domestic pharmaceutical company is consistently coming up as a potential joint sales partner for Mounjaro. There is speculation that the product may be launched under Lilly directly, then potentially transition to joint sales in the future. The company plans to actively promote Mounjaro's higher weight loss efficacy compared to Wegovy. Mounjaro demonstrated superior weight loss efficacy in the SURMOUNT-5 clinical trial, a head-to-head trial that compared Mounjaro with Wegovy. In the trial, the average weight loss rate at week 72 in the Mounjaro treatment group (10mg or 15mg) was 20.2%, compared to 13.7% in the Wegovy treatment group (1.7mg or 2.4mg), demonstrating a relatively improved weight loss rate. There is also high anticipation in the medical community. A hospital search application is promoting “how to book the lowest-priced hospital for Mounjaro.” Numerous posts promoting advance reservations for Mounjaro are also searchable on major portals and social media platforms. A representative from Lilly Korea stated, “Based on our global experience with the launch of Mounjaro in various countries and our deep understanding of the domestic market accumulated over more than 40 years, we will do our best to help patients with diabetes and obesity in Korea lead better lives with Mounjaro.” Wegovy’s domestic supply price reduced by up to 40% in response...Possibility of joint sales with Chong Kun Dang Ahead of the official launch of Wegovy, Novo Nordisk has responded with a price reduction for Wegovy. Novo Nordisk announced that it will reduce the price of Wegovy by up to 40% starting from the 14th. Excluding the highest dosage of 2.4mg, the prices will be reduced by 40% for 0.25mg, 30% for 0.5mg, 20% for 1.0mg, and 10% for 1.7mg. The price of 0.25mg, which will be reduced by 40%, is expected to be between KRW 200,000 and 220,000. This is analyzed as securing price competitiveness compared to Mounjaro. The supply prices for Mounjaro are ▲2.5 mg KRW 278,066, ▲5 mg KRW 369,307, and ▲7.5 mg and 10 mg KRW 521,377. The 2.5 mg and 5 mg doses will be initially supplied to Korea. The possibility of joint sales with Chong Kun Dang continues to be raised. Its possibility of partnering with Chong Kun Dang for Wegovy’s sales has also been consistently raised. Novo Nordisk and Chong Kun Dang have been in discussions regarding joint sales of Wegovy up until recently, and it is reported that they are nearing a final decision. However, both companies have drawn the line, saying that “nothing has been finalized yet.” If Chong Kun Dang is confirmed as a co-promoter for Wegovy and Mounjaro also signs a contract with a domestic pharmaceutical company, the competitive landscape is expected to expand from a head-to-head battle between two multinational pharmaceutical companies to include domestic pharmaceutical companies as well. The pharmaceutical industry is closely watching how quickly Mounjaro can catch up to Wegovy, which has effectively dominated the market. Wegovy quickly dominated the obesity treatment market after since launch in Korea in October last year. According to pharmaceutical market research firm IQVIA, Wegovy recorded sales of KRW 79.4 billion in the first quarter of this year. Wegovy's market share in the overall obesity drug market reached 73.2%. Wegovy surged to the top of the obesity drug market with sales of KRW 60.3 billion in the fourth quarter of last year, immediately after its launch. Wegovy has also contributed to the expansion of the market size. Prior to its launch in the third quarter of last year, the domestic obesity treatment market size was only KRW 47.4 billion, but it expanded to KRW 93.8 billion in the fourth quarter of last year, nearly doubling in just over three months.

- Company
- "Winrevair, gamechanger for pulmonary arterial hypertension"
- by Whang, byung-woo Aug 18, 2025 06:01am
- Winrevair (sotatercept), a new mechanism treatment for pulmonary arterial hypertension that has emerged after over 20 years, has been approved in Korea. Consequently, competitive market is expected. Although reimbursement remains a challenge, it is expected to change the treatment paradigm for pulmonary hypertension, a disease with significant unmet needs, by addressing the fundamental cause of the disease. Dr. Wook-Jin Chung, President of the Korean Society of Pulmonary HypertensionMSD Korea held a press conference on August 12, celebrating the approval of Winrevair and highlighting the drug's role and value. Pulmonary hypertension is not simply a state of constricted blood vessels. It is a disease of structural narrowing where the walls of the pulmonary arterioles thicken and their lumens become constricted, making it different from general hypertension. Dr. Wook-Jin Chung, President of the Korean Society of Pulmonary Hypertension (Professor of Cardiology at Gachon University Gil Medical Center), explained, "Pulmonary hypertension patients suffer from shortness of breath, cough, fatigue, and fainting as the disease advances. However, due to a complex diagnostic process, patients experience a diagnostic odyssey that can take up to three years." "Pulmonary hypertension patients experience such severe shortness of breath that even walking becomes difficult, leading to serious limitations in all aspects of daily life, including exercise, childcare, housework, and social activities. In severe cases, it can be fatal, with a risk of sudden death." For high-risk patients, the probability of death within one year exceeds 20%, and approximately 30% of domestic patients still die within five years of diagnosis. Until now, phosphodiesterase-5 (PDE5) inhibitors and calcium channel blockers (CCBs) have been used, but they have only been effective in controlling symptoms. Dr. Chung said, "Pulmonary hypertension is both a rare, intractable disease and a chronic condition, so there was an urgent need for a treatment strategy that could restore a healthy daily life to patients." In relation to this, the newly approved Winrevair is generating high expectations because it approaches the fundamental cause of the disease. Winrevair is the first-in-class Activin Signaling Inhibitor (ASI), utilizing the 'Activin' pathway, which is one of the pathogenic mechanisms of pulmonary hypertension. This treatment demonstrates the potential for reverse remodeling by inhibiting the proliferation of abnormally thickened blood vessels, thereby reversing the remodeling of pulmonary arterioles and the right ventricle. In a study of 323 pulmonary hypertension patients, the Winrevair group's 6-minute walk distance (6MWD) increased by 40.1 m, while the placebo group's decreased by 1.4 m. Additionally, Winrevair reduced the risk of clinical worsening or death from all causes related to pulmonary hypertension by 84%. Kyung-hee Kim, Chairperson of the Clinical Guidelines Committee of the Korean Society of Pulmonary Hypertension, said, "Experts from the clinical guidelines committees in Europe and the United States have recommended the addition of ASI when updating pulmonary hypertension treatment guidelines," and added, "The Korean Society of Pulmonary Hypertension is also reviewing the inclusion of ASI in the pulmonary hypertension treatment guidelines in Korea." Dr. Kyung-hee Kim, Chairperson of the Clinical Guidelines Committee of the Korean Society of Pulmonary HypertensionWinrevair was approved through the Ministry of Food and Drug Safety's 'Concurrent Approval-Evaluation-Negotiation Pilot Program' and has quickly entered the reimbursement review process. The Ministry of Health and Welfare (MOHW) has been running the 'Concurrent Approval-Evaluation-Negotiation Pilot Program' since 2023 to improve access to treatment for life-threatening, severe, and rare diseases. The project conducts approval, reimbursement evaluation, and drug price negotiations simultaneously, aiming to shorten the time required for new drugs to be included in the National Health Insurance list. Dr. Chung emphasized, "For patients to benefit from Winrevair, it must be quickly covered by national health insurance." He added, "Even if the treatment is expensive, and its efficacy has been proven overseas, efforts are necessary to ensure it can be used, even if strict criteria are applied."
- Company
- Imfinzi denied reimb for bile duct cancer for the 3rd year
- by Eo, Yun-Ho Aug 14, 2025 06:13am
- Attention is focused on whether there will be progress in the reimbursement process for Imfinzi for bile duct cancer, which has remained non-reimbursed for three years now. According to industry sources, the Health Insurance Review and Assessement Service's Drug Reimbursement Evaluation Committee may likely review the agenda of expanding the reimbursement of AstraZeneca Korea's PD-L1 inhibitor immunotherapy Imfinzi (durvalumab) for bile duct cancer in the second half of the year. In the harsh environment of bile duct cancer, where the average survival period was just over seven months, the emergence of Imfinzi brought about a change in the treatment landscape. Imfinzi demonstrated more than a twofold improvement in overall survival rate compared to the standard chemotherapy regimen at the three-year mark, and in a subgroup analysis of Korean patients, it proved to provide even better survival benefits than the overall patient population. Countries such as Canada, the United Kingdom, Australia, Japan, and Taiwan quickly reimbursed Imfinzi in recognition of its clinical innovation. In the case of the United Kingdom, considering the poor reality of bile duct cancer treatment and the fact that Imfinzi is the first first-line treatment for bile duct cancer, the ICER threshold was applied flexibly by exceptionally weighting the quality-adjusted life years (QALY) during the pharmacoeconomic evaluation process. However, in South Korea, reimbursement barriers remain, limiting patients' access to treatment. It has been three years since Imfinzi was approved by the Ministry of Food and Drug Safety as a first-line treatment for metastatic bile duct cancer. The indication for bile duct cancer was approved by the Cancer Review Committee in November 2024, but there has been no significant progress to date. Jung-yong Hong, Professor of Hematology and Oncology at Samsung Medical Center, said, “The ultimate goal of the government, pharmaceutical companies, and medical professionals is to provide Imfinzi’s treatment benefits, proven as standard therapy, to bile duct cancer patients as quickly as possible. Measures to strengthen institutional flexibility must be explored to enhance treatment access for bile duct cancer patients.”