- LOGIN
- MemberShip
- 2026-06-12 16:51:28
- Company
- Lotte Biologics·SK Pharmteco jointly target global ADC mkt
- by Cha, Jihyun Oct 31, 2025 06:12am
- Letter of Intent signing ceremony between Lotte Biologics and SK Pharmteco (Source: Lotte Biologics) Lotte Biologics (CEO James Park) and SK Pharmteco (CEO Joerg Ahlgrimm) announced on the 30th that they have signed a Letter of Intent (LOI) for strategic business collaboration to strengthen their competitiveness in the global Antibody-Drug Conjugate (ADC) market. The signing ceremony took place at the Lotte Biologics booth at ‘CPHI Worldwide 2025’, the world's largest pharmaceutical and biotech industry held in Frankfurt, Germany. Executives from both companies, including James Park, CEO of Lotte Biologics; Yoo-Yeol Shin, Head of Global Strategy; Joerg Ahlgrimm, CEO of SK Pharmteco; and Andrew Penny, CCO. Through the collaboration, the two companies plan to jointly provide an integrated, one-stop CDMO service based on various ADC-specialized solutions to potential customers in the global market. Lotte Biologics will leverage its cGMP manufacturing capabilities and global quality competitiveness at the Syracuse Bio Campus in the U.S. to provide ADC-specific CDMO services from API manufacturing to conjugation. SK Pharmteco will be responsible for the chemical synthesis processes, including linkers and payloads. By optimizing and combining each company’s respective expertise, the companies aim to establish a full-cycle CDMO system, deliver customized integrated services, expand order intake through joint marketing, and secure global clients through joint marketing. Furthermore, they aim to address the clients’ overall chemical, manufacturing, and control (CMC) requirements and present an integrated technology and operational platform that minimizes gaps between development and manufacturing. Lotte Biologics expects to provide differentiated value to customers based on its credibility and quality competitiveness as a global bio CDMO. This is underpinned by its antibody conjugation production infrastructure at its Syracuse, USA facility, global client service experience, and track record of flawless FDA inspections. Lotte Biologics further explained that this agreement, leveraging the synergies of the production infrastructures in the U.S. and Europe, will also enable a strategic response to the ongoing US-driven supply chain restructuring and rising bio-reshoring demands. James Park, CEO of Lotte Biologics, said, “This collaboration represents a significant achievement as the pharmaceutical-biotech affiliates of Korea's leading companies join forces on the global stage for the first time. We will strive to secure a competitive advantage in next-generation modalities such as ADCs and demonstrate the potential of K-Bio on the global stage.” Joerg Ahlgrimm, CEO of SK Pharmteco, said, “This strategic partnership will accelerate the development of next-generation therapy, enabling both companies to deliver greater value and bring innovative new drugs to patients around the world faster.”
- Policy
- Reimb expanded for Opdivo, Yervoy…set for Vyloy, Padcev
- by Jung, Heung-Jun Oct 31, 2025 06:11am
- BMS Korea’s Yervoy (ipilimumab) and Ono Pharmaceutical Korea’s Opdivo (nivolumab) passed their first hurdle toward expanding reimbursement in Korea. Meanwhile, Janssen Korea’s Tecvayli (teclistamab), Pfizer Korea’s Elrexfio (elranatamab), and Astellas Korea’s Padcev (enfortumab vedotin) and Vyloy (zolbetuximab) had their reimbursement standards established, clearing the first hurdle toward reimbursement listing. The Health Insurance Review and Assessment Service (HIRA) held its 8th Cancer Disease Deliberation Committee meeting for 2025 on the 29th to deliberate on reimbursement criteria for anticancer drugs. Among these, Astellas Pharma Korea saw a double win, as reimbursement criteria were set simultaneously for two of its products. In the case of Vyloy, the National Assembly had requested expediting its reimbursement decision process. In a written inquiry during the Ministry of Health and Welfare's National Assembly audit. This time, Vyloy’s reimbursement criteria were established for ‘First-line treatment for patients with unresectable locally advanced or metastatic gastric or gastroesophageal junction adenocarcinoma who are CLDN18.2-positive and HER2-negative’ For Padcev, reimbursement criteria were set for both combination therapy and monotherapy, as ‘combination therapy with pembrolizumab as first-line treatment for adult patients with locally advanced or metastatic urothelial carcinoma’ and ‘monotherapy for adult patients with locally advanced or metastatic urothelial carcinoma who have previously received treatment with a PD-1 or PD-L1 inhibitor and platinum-based chemotherapy.’ In addition, Pfizer Korea’s Elrexfio (elranatamab) and Janssen Korea’s Tecvayli (teclistamab) also had their reimbursement criteria established, clearing their first hurdle for listing. For Opdivo, which sought to expand the scope of reimbursement, criteria were set for its use in combination therapy as first-line treatment for metastatic esophageal squamous cell carcinoma. The reimbursement criteria have also been established for the Opdivo and Yervoy combination, but only for specific patient populations. It was granted reimbursement for ‘first-line treatment of adult patients with unresectable malignant pleural mesothelioma,’ but not for hepatocellular carcinoma or non-small cell lung cancer.

- Company
- "Severe COVID-19 cases continue to rise...infection control"
- by Son, Hyung Min Oct 30, 2025 06:11am
- Joon Young Song, Professor of Infectious Diseases at Korea University Guro HospitalMore than two years after the COVID-19 pandemic transitioned to an endemic phase, the number of severe cases, particularly among high-risk groups, continues to occur steadily. 'The shadow of infection' remains in clinical practice. While the number of confirmed cases is not surging as before, hospitalizations and severe cases are persisting, especially among vulnerable populations. Joon Young Song, Professor of the Department of Infectious Diseases at Korea University Guro Hospital, said, "Many high-risk patients hospitalized with severe symptoms like pneumonia are confirmed with COVID-19." He added, "These patients face risks of prolonged hospitalization, deterioration of physical function, and death from severe infection." Professor Song also said, "Most patients hospitalized for pneumonia are elderly or immunocompromised, both high-risk groups. Since recovery is slow and the possibility of complications is high, prevention through vaccination is essential." According to Professor Song, age is the most significant risk factor for COVID-19, with the risk of progression to a severe case increasing significantly with age in those 65 and older. Immunocompromised individuals are at a very high risk of severe infection. An analysis of vaccine efficacy and disease burden, conducted in the 'Host-based Influenza Morbidity & Mortality Study' involving eight medical institutions, including Korea University Guro Hospital, showed a high risk of COVID-19 infection and severe disease among immunocompromised patients, such as cancer patients and chronic kidney disease patients. Dialysis patients are also classified as high-risk. Since hospital dialysis rooms are confined spaces where multiple patients receive treatment, an infection can rapidly spread into an outbreak, with most cases carrying a high risk of progression to severe infection. Experts are increasingly viewing COVID-19 as a seasonal respiratory illness, actively recommending vaccination and raising awareness of the disease. Professor Song said, "In recent years, COVID-19 has shown a pattern of recurrent outbreaks twice a year, during summer and winter," and stressed, "The severe risk must be lowered through regular vaccination, just like with the flu." However, public awareness of the importance of vaccination has decreased as the endemic transition progresses. According to Professor Song, even patients who absolutely need the vaccine, such as cancer patients, often hesitate due to concerns like 'Will I get a fever?' or 'Will it interfere with my anti-cancer treatment?' if their physician does not absolutely recommend it. Professor Song emphasized that if a cancer patient contracts COVID-19 without being vaccinated, there is a high probability that their anti-cancer treatment must be suspended or that the infection will progress to a severe illness, such as pneumonia. Professor Song assessed, "The preventive effect against severe infection remains over 70% and is maintained for about 10 months after vaccination. For high-risk groups, such as the older adults aged 65 and over or those with underlying conditions like heart failure or lung disease, the priority is reducing hospitalization and death resulting from severe infection, pneumonia, acute respiratory illness, and the worsening of underlying diseases, rather than just preventing the infection itself." He also emphasized, "It is crucial for medical professionals to fully explain the necessity and safety of vaccination to patients and actively recommend it." "Safety of vaccination against COVID-19·flu at the same time, and the need to boost coverage" The government began the National Immunization Program (NIP) for both flu and COVID-19 vaccines on October 15. Simultaneous vaccination of the two is possible. This year's vaccine strain is LP.8.1 strain, a variant that began circulating last year. Recently, sub-variants that have evolved from LP.8.1 strain, such as NB.1.8.1 strain and XFG strain, have been increasing. Experts assess that, since these variants are sublineages of the Omicron lineage, the LP.8.1 variant vaccine provides sufficient protection. Immunogenicity evaluation has confirmed a high level of cross-immunity against these sub-variants with the LP.8.1 vaccine. Professor Song said, "As COVID-19 repeats its annual outbreaks, it is significant that the government has officially decided to support vaccination for the age groups and patient populations who absolutely require it." Pfizer received approval for its 'Comirnaty LP.8.1' vaccine targeting the LP.8.1 variant in August. Notably, Pfizervaccine comes in a pre-filled syringe (PFS) formulation. While multi-dose vials may have slight dosage errors, PFS offers the advantage of administering the correct, predetermined dose in a single-use syringe. PFS is also considered safer in terms of infection control. Professor Song commented, "The latest COVID-19 vaccines have confirmed sufficient preventive efficacy. The currently circulating variant viruses do not have large mutational differences, so the vaccines introduced domestically can be expected to provide sufficient protection against the latest variants." He added, "Like the flu vaccine, the COVID-19 vaccine requires regular administration. It is difficult to say which of the two diseases is more severe definitively, but both COVID-19 and influenza pose a high disease burden for those aged 65 and older." However, the COVID-19 vaccine initially had a lower vaccination rate than the flu vaccine, due to a perception that it was difficult to tolerate and caused many side effects. While the influenza vaccination rate for those aged 65 and older currently reaches about 85%, the COVID-19 vaccination rate was 45% at the end of last year and 47.9% as of April this year. Therefore, recommending concurrent vaccination of both is necessary to boost COVID-19 vaccination rates. Professor Song explained, "The events after COVID-19 vaccine administration are a condition resulting from immune boosting. However, from the third dose onward, the frequency and intensity of general adverse events, such as fever and fatigue, have decreased. The incidence of myocarditis, which was a concern among young people, has also decreased significantly. We believe that these concerns will gradually improve as COVID-19 vaccines are administered repeatedly." He also expressed that the safety of concurrent vaccination is not an issue. He said, "Concurrent vaccination has not increased the frequency of adverse events, and no severe cases have been reported. Various studies have all confirmed that simultaneous vaccination with the COVID-19 vaccine and the influenza vaccine is safe." Professor Song added, "Simultaneous vaccination plays an important role in increasing vaccination rates. Administering both vaccines during a single visit prevents patients from missing their vaccination window. In fact, a significant number of patients who received the COVID-19 vaccine did so simultaneously with the influenza vaccine." Finally, Professor Song said, "Academic societies are cooperating with the KDCA to continue communicating the necessity of the COVID-19 vaccine through various media and are continuing activities like campaigns." He stressed, "Misinformation or distorted reports related to vaccines, once spread, are often perceived as facts by the public and are difficult to correct. Therefore, actively sharing accurate information and continuously educating and promoting the safety and necessity of vaccines is important."

- Policy
- K-API·listed essential medicine to get preferential price
- by Lee, Jeong-Hwan Oct 30, 2025 06:11am
- Minister Jeong Eun Kyung promised Rep. Baek Jong-heon to implement the The Ministry of Health and Welfare (MOHW) has decided to expand eligibility for the 68% preferential drug price for National Essential Medicines that use domestic active pharmaceutical ingredients (API). The MOHW aims to foster the domestic industry by incentivizing the use of domestically sourced API. The MOHW is shifting its initial stance—which applied the 68% preferential drug price only to essential medicines that received marketing authorization after the policy's implementation in March of this year—to granting the 68% preferential price to already listed products. Notably, the MOHW will also add the measure of domestic API self-sufficiency to its ongoing research on advanced policies for drugs with unstable supply. Furthermore, it plans to review the need for a preferential drug price for combination drugs using domestic API at a later stage. This is a follow-up measure by the MOHW after Rep. Baek Jong-heon (People Power Party) criticized Minister Jeong Eun Kyung during a parliamentary inspection this year, pointing out a policy failure: not a single pharmaceutical company had applied for or benefited from the 68% preferential prices of essential medicines. This announcement is from the 'Roadmap for Fostering Domestic Active Pharmaceutical Ingredients' submitted by the MOHW to Rep. Baek Jong-heon's office on October 29. According to the documents submitted to the representative's office, the MOHW will expand the eligibility for the 68% domestic API preferential drug price, currently applied only to newly listed National Essential Medicines, to include 'already listed essential medicines.' To establish this, the MOHW stated that it initiated a survey on the status of domestic API-using drugs held by domestic pharmaceutical companies on the 20th, through organizations such as the Korea Pharmaceutical and Bio-Pharma Manufacturers Association and the Korea Biomedicine Industry Association. Rep. Baek explained that, based on the survey results, essential medicines that were already listed and had obtained National Health Insurance drug prices before the implementation of the preferential policy in March are expected to receive a retroactive application of the 68% price hike. The MOHW also decided to include strategies for API self-sufficiency in the ongoing research service to analyze trends and stabilize the supply of drugs with unstable supply (from September to November 2025). Upon completion of this research, the ministry will consider commissioning additional research to develop an API self-sufficiency roadmap. The MOHW plans to increase the Korea Health Industry Development Institute (KHIDI) budget by KRW 100 million next year for this purpose. The MOHW stated, "Next year's project to strengthen API self-sufficiency will address the excessive reliance on specific countries for imported APIs, such as China (35%) and India (15%)," and stressed, "We are also pursuing the expansion of the 68% preferential drug price for National Essential Medicines using domestic API to include already listed essential medicines. We are currently confirming the status and gathering opinions through the pharmaceutical and biotech organizations."
- Company
- 'Januvia generics hit 23% market share in 2 years'
- by Kim, Jin-Gu Oct 30, 2025 06:11am
- Generic versions of the DPP-4 inhibitor class diabetes treatment ‘Januvia (sitagliptin)’ series expanded their market share to 23% just two years after launch. However, analysis indicates their market penetration pace is slower than that of previous cases such as Tenelia (teneligliptin) and Galvus (vildagliptin). Performance among generic manufacturers varied significantly. While most generics recorded quarterly prescription sales below KRW 100 million, Kyongbo Pharmaceutical and Hanmi Pharmaceutical achieved results exceeding KRW 1 billion, strengthening their presence. Januvia generics hit 23% share in 2 years — market penetration slower According to the pharmaceutical market research institution UBIST on the 29th, total outpatient prescriptions for sitagliptin monotherapy and sitagliptin/metformin combinations in Q3 this year reached KRW 28.3 billion, down 6% from KRW 30.2 billion in the same quarter last year. While the sales of original products declined, generics grew. Sales of the original Januvia, Janumet, and Janumet XR fell 14%, from KRW 25.1 billion in Q3 2023 to KRW 21.7 billion this year. Specifically, Januvia dropped from KRW 5.8 billion to KRW 4.5 billion, and sales of Janumet/Janumet XR combined declined from KRW 19.3 billion to KRW 17.2 billion. Since the launch of the generics, the originals have seen a steady downward trend. Just before patent expiry in Q2 2023, total prescriptions for the Januvia series stood at KRW 37.5 billion, but after generics entered in Q4 2023, the figure fell to KRW 26.6 billion, then dropped further 18% by Q3 this year. Meanwhile, generic products saw their combined prescription sales increase by 30% over the past year, rising from KRW 5.1 billion to KRW 6.6 billion. Sales of Januvia generics increased from KRW 1.6 billion to KRW 1.9 billion, while Janumet and Janumet XR generics rose from KRW 3.5 billion to KRW 4.7 billion. Januvia generics were released in September 2023. Following the patent expiration of the long-standing market leader of the KRW 600 billion annual DPP-4 inhibitor market, many generic companies rushed to join in the competition. A total of 89 companies received approval for generics, and 60 of these companies launched products. The overall market share of generics expanded from 17% to 23% within a year. However, the pace of the market penetration is somewhat slower compared to other DPP-4 inhibitor generics like Tenelia and Galvus. Galvus and Galvusmet generics recorded a 42% market share in their first year. In its second year, it further expanded its share to 47%, reaching a level similar to the original. Tenelia and Tenelia M generics surpassed the original with a 54% market share in their first year. This rose to 58% in their second year. Sales of Hanmi·Kyongbo soar... Most other companies record quarterly prescription sales under KRW 300 million Generic companies showed polarized results. Only two companies recorded quarterly prescription sales exceeding KRW 1 billion, while most others fell short of even KRW 100 million. Kyongbo Pharmaceutical reported KRW 1.1 billion in Q3 sales for its combination generics Janustinmet and Janustin XR, nearly doubling from KRW 600 million in the same quarter last year. Cumulative prescriptions for the two products have reached KRW 4.9 billion. Hanmi Pharmaceutical’s Sita and Sitamet XR posted KRW 1 billion, up 25% year-over-year, bringing cumulative prescriptions to KRW 5.8 billion since launch. In contrast, most generics are underperforming. Of the 60 companies that launched products, 41 (68%) posted quarterly sales below KRW 100 million, and 15 companies (25%) recorded less than KRW 300 million. The average Q3 prescription per company stood at approximately KRW 110 million. The reason Januvia generics are struggling to expand their influence is that the DPP-4 inhibitor class diabetes treatment market is already saturated. In this space, Januvia competes with original products such as Zemiglo (gemigliptin), Trajenta (linagliptin), Tenelia (teneligliptin), Suganon (evogliptin), Galvus (vildagliptin), Onglyza (saxagliptin), Nesina (alogliptin), and Guardlet (alogliptin/metformin). Among these, Galvus, Tenelia, and Trajenta have already faced generic competition upon their patent expiry. Furthermore, the DPP-4 inhibitor market has seen a decline in overall prescription volume since the emergence of SGLT-2 inhibitor class diabetes treatments like Forxiga and Jardiance. In this environment, the flood of Januvia and Janumet generics has led to fierce competition and lower-than-expected sales performance.
- Company
- Wegovy prescribed to adolescents for obesity
- by Son, Hyung Min Oct 30, 2025 06:09am
- Novo Nordisk held an event on the 28th at the Courtyard by Marriott Seoul Namdaemun for its obesity treatment drug Wegovy, which has settled as a leading therapy for adult obesity, has expanded its reach into adolescent obesity. Following approvals in the US and Europe, Novo Nordisk’s Wegovy can now be prescribed to patients aged 12 and older in Korea. With this, calls from the medical community on earlier intervention for obesity are gaining traction. Experts describe this approval as “not merely an expansion of indication, but an institutional turning point recognizing adolescent obesity as a chronic disease.” Adolexcent obesity moves out of the treatment blind spot Novo Nordisk held a press conference on the 28th to mark the expanded adolescent indication for its obesity treatment Wegovy (semaglutide 2.4mg), sharing key clinical results and future treatment strategies. Wegovy is a once-weekly GLP-1 (glucagon-like peptide-1) receptor agonist approved last year in Korea for treating adult obesity. With the new indication, it can now be used for adolescent patients aged 12 and above in Korea, emerging as a new treatment option for obesity during the growth years. According to Novo Nordisk, semaglutide 2.4mg is structurally similar to human GLP-1. It regulates overall metabolism not only through weight loss but also by improving blood glucose and lipid levels. Ju Ok Lim, Head of Medical Affairs at Novo Nordisk, said, “Semaglutide 2.4mg demonstrated consistent weight loss effects and safety not only in adults but also in adolescents. The indication expansion will serve as an opportunity to address the treatment gap for adolescent obesity.” Until now, obesity treatments available for children and adolescents aged 12 and older in Korea have been extremely limited. Representative options include the fat absorption inhibitor ‘orlistat’ and the once-daily GLP-1 class injectable ‘Saxenda (liraglutide)’. Latest adult-targeted medications like ‘Qsymia (phentermine/topiramate)’ and ‘Mounjaro (tirzepatide)’ are not yet approved for adolescent use. The approval of Wegovy expands access to weekly therapy-based treatment approaches. Julie Broe Honore, Head of clinical development, medical and regulatory affairs at Novo Nordisk, said, “Adolescent obesity is no longer an individual problem but a public health issue. This expansion of Wegovy's indication marks the first step toward preventing adult metabolic diseases through early intervention.” STEP TEENS study…16% weight reduction & improved cardiovascular risk factors This expansion of Wegovy's indication is based on results from the Phase III STEP TEENS clinical trial conducted in adolescents. The study involved 201 adolescents aged 12-18 who were obese or overweight and had weight-related conditions, conducted over 68 weeks. The average age was 15.4 years, 62% were female, and the racial distribution was 79% White, 8% Black, and 2% Asian. Clinical results showed the Wegovy group achieved an average BMI (Body Mass Index) reduction of 16.1%, demonstrating a significant difference compared to the placebo group, which increased by 0.6%. Approximately 72.5% of patients achieved at least a 5% weight loss, nearly four times higher than the placebo group (17.7%), with an average weight loss of 15.3 kg. Notably, 37.4% of patients achieved a weight loss of 20% or more, demonstrating efficacy in adolescents comparable to that seen in adults. Improvements were observed not only in cardiovascular risk factors like waist circumference, blood pressure, and lipid levels, but also in quality of life (QoL) indicators. Eun-gu Kang, Professor of Pediatrics at Korea University Ansan HospitalThe main adverse reactions reported were gastrointestinal symptoms such as nausea, vomiting, and diarrhea, but most were mild and did not affect growth or pubertal development. Eun-gu Kang, Professor of Pediatrics at Korea University Ansan Hospital, commented, “Beyond weight reduction, the study confirmed metabolic benefits such as LDL cholesterol reduction and improvements in blood pressure and waist circumference, suggesting positive cardiovascular implications.” He further noted, “However, as adolescents are still in the process of growth, long-term follow-up and accumulation of safety data are necessary.” Adolescent obesity rate increases 1.7 times over 10 years Experts unanimously agreed that this approval holds significance in terms of ‘securing treatment accessibility’. According to the ‘2024 Adolescent Health Behavior Survey’ conducted by the Korea Disease Control and Prevention Agency and the Ministry of Education, the obesity rate among middle and high school students rose from 7.5% in 2015 to 12.5% last year, a 1.7-fold increase over 10 years. The overweight/obesity rate for male students is 43.0%, and for female students, 24.6%, the highest among the four East Asian countries, including China, Japan, and Taiwan. The main concern is comorbidities. Around 80% of obese adolescents remain obese into adulthood and often develop one or more metabolic disorders, such as hypertension, diabetes, or dyslipidemia. Yong-hee Hong, Professor of Pediatrics at Soonchunhyang University Bucheon HospitalThis is why experts point out that adolescent obesity should be viewed not as a mere ‘cosmetic issue’ but as a disease requiring early intervention. Yong-hee Hong, Professor of Pediatrics at Soonchunhyang University Bucheon Hospital, emphasized “Obesity is clearly a disease requiring treatment, yet it is often still perceived as a cosmetic issue. This perception must change so that patients who actually need medication can be brought into the healthcare system.” Young-sung Suh, Professor of Family Medicine at Keimyung University Dongsan Hospital and President of the Korean Society for the Study of Obesity, stated, “Obesity is not a matter of willpower but a complex metabolic disorder. At the society level, we will focus on enhancing treatment accessibility while ensuring safe medication use and preventing misuse.” While some raise concerns about muscle loss with GLP-1 agonists, experts see significant clinical benefits due to their fat-focused weight reduction. Lim said, “Some muscle loss may occur with weight reduction, but overall, the health benefits from fat loss are far greater. We will strengthen education and management to enable healthy weight loss alongside the metabolic improvement.” Kang added, “While muscle loss in patients during growth periods requires caution, GLP-1 drugs often yield positive outcomes in body composition changes. Controlling fat mass and maintaining nutritional balance are more important than just body weight numbers.”
- Policy
- Jardiance generics listed late October…but not CKD
- by Jung, Heung-Jun Oct 30, 2025 06:09am
- As the substance patent for Jardiance expired on October 24, a total of 235 products—both single-agent and combination formulations—were simultaneously added to Korea’s reimbursement list. However, Chong Kun Dang chose a different approach, applying for sequential listing of its single-agent and combination products. The company first listed its single-agent Empamax Tab (10 mg, 25 mg) on the reimbursement list in line with the patent expiration date this month and plans to add 13 combination formulations in November. According to industry sources on the 29th, unlike other generic manufacturers, Chong Kun Dang owns a salt-modified single-agent formulation, which led to this stepwise, sequential reimbursement strategy. Chong Kun Dang’s Empamax Tab (10 mg, 25 mg) contains empagliflozin L-proline. It is a salt-modified version of the original compound, unlike other generics. By conjugating L-proline, an amino acid, the company successfully avoided patent infringement. After review by the Health Insurance Policy Deliberation Committee, the products have been reimbursed since October 24 at the same price as their planned sales price. Starting in November, 13 additional combination formulations—both dual and triple combinations—will be listed. A total of 11 products, that are combinations of empagliflozin and metformin-Empamax XR Tab (10/1000 mg, 12.5/1000 mg, 25/1000 mg), Empamax M Tab (12.5/500 mg, 12.5/850 mg, 12.5/1000 mg, 5/500 mg, 5/850 mg, 5/1000 mg), and Empamax S Tab (10/100 mg, 25/100 mg- will be granted reimbursement. In addition, two triple-combination formulations containing empagliflozin + sitagliptin + metformin—marketed as Emsiformin XR Tab—will also be added. A Chong Kun Dang official explained, “We modified the salt form to enhance safety. Unlike other generics, we first listed the single-agent product, and our combination products are being reimbursed subsequently based on the single-agent drug data.” Chong Kun Dang has received marketing approval for a total of 18 products, including both single-agent and combination versions, but has submitted reimbursement applications for only 15. Some triple-combination formulations and specific dosages were not included in the application. The company plans to prioritize products with higher prescription potential. The official added, “We are first launching the dosages expected to have higher prescription volume, and will apply for additional listings depending on market conditions.”
- Company
- Reimb for Perjeta as adjuvant therapy under CDDC review
- by Eo, Yun-Ho Oct 29, 2025 06:12am
- The expansion of reimbursement coverage for Perjeta’s use as adjuvant therapy in breast cancer is drawing attention. According to industry sources, Roche Korea’s HER2-positive breast cancer treatment Perjeta (pertuzumab) has been submitted for review by the Health Insurance Review and Assessment Service (HIRA) Cancer Disease Deliberation Committee. Currently, Perjeta is reimbursed for treating HER2-positive metastatic or unresectable locally recurrent breast cancer. In early breast cancer, it is granted selective reimbursement as a neoadjuvant therapy (before surgery), with patients bearing 30% of the cost. However, in the postoperative adjuvant therapy stage—a key treatment step to prevent recurrence—has remained non-reimbursed (100% patient coinsurance) since its indication was added in Korea in 2018, limiting patient access. The recent submission to the National Health Insurance Service's Drug Disease Deliberation Committee is believed to have been significantly influenced by the 10-year long-term follow-up data from the Phase III APHINITY clinical trial, presented at the European Society for Medical Oncology Breast Cancer Conference (ESMO Breast Cancer 2025) last May. This study newly confirmed statistically significant final overall survival (OS) data for the combination of Perjeta and Herceptin (trastuzumab). When Perjeta was administered in combination with Herceptin and chemotherapy, the risk of death in patients with HER2-positive early breast cancer was reduced by 17% compared to the existing Herceptin-chemotherapy combination therapy. Furthermore, the 10-year survival rate in the Perjeta-Herceptin treatment arm was 91.6%, showing an improvement compared to the 89.8% in the control arm. The Perjeta-Herceptin treatment arm also demonstrated greater clinical benefit in patients at high risk of recurrence. Specifically, in a subgroup analysis of patients with lymph node–positive disease, who are at high risk of recurrence, the combination showed a 21% reduction in the risk of death. Furthermore, the benefit in invasive disease-free survival (iDFS) was also maintained, reaffirming the earlier findings of the APHINITY study. It remains to be seen whether Perjeta's adjuvant therapy indication can pass CDDC review and ultimately be granted reimbursement coverage. Meanwhile, the Perjeta and Herceptin combination is currently recommended as Category 1 adjuvant therapy in the U.S. NCCN Guidelines for patients with HER2-positive early breast cancer and lymph node metastasis. It is also recommended as Category 1 adjuvant therapy for high-risk, lymph node–positive patients who achieved pathological complete response (pCR) after neoadjuvant chemotherapy.
- Company
- Will the Indian API cause problems?
- by Kim, Jin-Gu Oct 29, 2025 06:12am
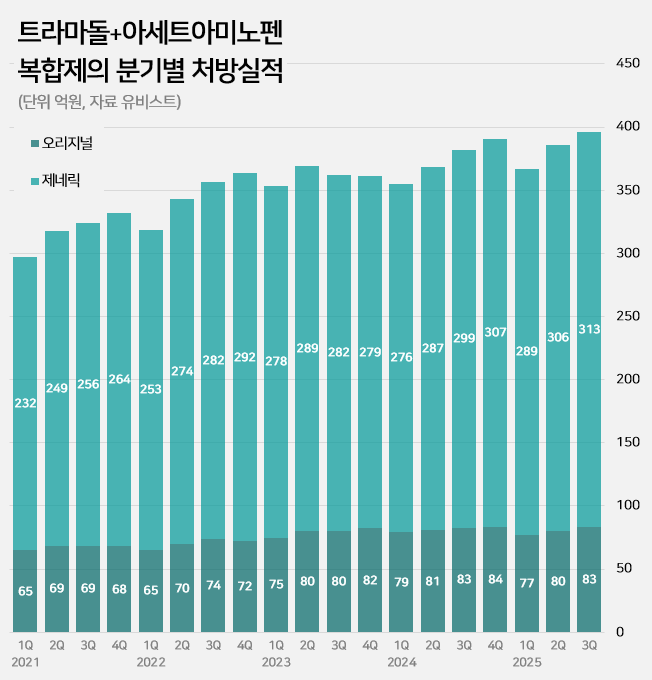
- An impurity issue has emerged as a major variable in the tramadol-acetaminophen combination drug market, valued at approximately KRW 150 billion annually. Following the detection of impurities in three generic products using Indian-sourced active pharmaceutical ingredients (API), the pharmaceutical industry is watching closely for the potential expansion of the impurity issue across all API from India. In this case, the market is expected to be reorganized around products that use non-Indian sourced API. 85% of tramadol DMFs are produced in India...Concerns Over Widespread Impurity Detection According to the Ministry of Food and Drug Safety (MFDS) on October 29, following the excess detection of the impurity NNDT (N-nitroso-desmethyl tramadol) in a tramadol monotherapy product in July, the recall target has recently been expanded to include tramadol + acetaminophen combination products. To date, impurities have been detected in three tramadol + acetaminophen combination products. The MFDS issued recall orders for specific batch number products of: ▲Dongkoo Bio & Pharma’s 'Jimuradol Tab' ▲Ausco Korea Pharma's 'Acetadol Tab' ▲Union Korea Pharm’s 'Atracen Tab.' All three products in which impurities were detected are known to have used India-sourced API. The concern is that a significant portion of the tramadol + acetaminophen combination products currently distributed in South Korea relies on API sourced from India. Currently registered sources of tramadol API: YELLO-India, BLUE-Israel, RED-Switzerland, GREEN-Korea Indeed, examining the Drug Master File DMF status for tramadol-related API shows that 62 of the 73 registered raw material manufacturers (85%) are located in India. The remaining 11 sites include 8 in Israel (11%), 2 in Switzerland (3%), and 1 in Korea (1%). Many domestically supplied products likely rely on Indian-sourced API. KRW 150 Billion Market...Top-selling products not at risk of impurity expected to benefit The three products where impurities were detected have negligible prescription sales. According to pharmaceutical market research firm UBIST, the cumulative prescription sales for these three products combined were less than KRW 400 million in the third quarter of this year. While the market impact is currently limited, analysis suggests that market restructuring will be unavoidable if impurity detection expands across Indian-sourced API. This is because securing new raw material manufacturers outside of India is difficult, and the administrative process for changing API registration alone takes approximately six months, which would lead to a market supply gap. The tramadol + acetaminophen combination market in Korea has seen steady growth in the moderate-to-severe pain treatment sector. It increased by 24% over the last four years, from KRW 120.7 billion in 2020 to KRW 127.1 billion in 2021, KRW 138.2 billion in 2022, KRW 144.6 billion in 2023, and KRW 149.6 billion last year. Cumulative sales reached KRW 114.9 billion in the third quarter of this year. Quarterly prescription sales of tramadol-acetaminophen combination drugs (DARK GREEN: original product, LIGHT GREEN: generic products; unit: KRW 100 million, source: UBIST). The original product, Janssen Korea's Ultracet, recorded cumulative sales of KRW 24.1 billion in the third quarter, a slight decrease from KRW 24.3 billion in the third quarter of last year. Generic products collectively saw a 5% increase in the same period, rising from KRW 86.3 billion to KRW 90.9 billion. The growth of top-selling generics was particularly notable: Samjin Pharm's 'Synerjet' grew by 23%, from KRW 6.9 billion to KRW 8.6 billion; Myung Moon's 'Traphen' grew from KRW 5.2 billion to KRW 6.6 billion; and Genuone Sciences' 'Painless' increased from KRW 4.4 billion to KRW 5.1 billion. In this circumstance, if the impurity issue spreads, non-detected products are expected to quickly replace the market. Currently, the market-leading products are understood to be not at risk. The original product, Ultracet, has registered a Swiss manufacturer as its API supplier. The top generic product, Samjin Pharm's Synerjet, manufactures its finished drug in-house using an Israeli-sourced API. Neither of these two products was requested to submit data in the MFDS's comprehensive investigation of NNDT impurities in tramadol.
- Policy
- Non-reimbursed approvals of off-label drugs
- by Jung, Heung-Jun Oct 29, 2025 06:10am
- The number of non-reimbursed approvals of off-label drugs: while 208 applications for off-label use were rejected, 2,186 were approved between 2020-2025.The number of approvals for non-reimbursed use of drugs beyond their authorized indications (off-label) has been more than 10 times the number of rejections over the past six years. While 208 applications for off-label use were rejected, 2,186 were approved, often because the benefits outweighed the risks. On October 27, the Health Insurance Review & Assessment Service (HIRA) publicly disclosed, for the first time, the number of non-reimbursed approvals for off-label drugs, including over-the-counter (OTC) medicines. Applications for non-reimbursed off-label drug use must first be reviewed by an IRB (Institutional Review Board) established in a hospital or by an academic society. For OTC medicines, HIRA commissions the Ministry of Food and Drug Safety (MFDS) to review safety and efficacy, while anti-cancer drugs are approved by the Cancer Disease Review Committee (CDRC), which holds monthly. Unlike anti-cancer drugs, only the records of rejections for OTC medicines had been disclosed until now. This had been criticized as a problem, making it difficult for healthcare institutions to file applications for off-label non-reimbursed use. There was also a persistent request to disclose approval records for OTC medicines to ensure fairness. HIRA pre-announced a partial amendment to the 'Partial Revision to Operational Regulations for Approval of Non-Reimbursed Off-Label Drug Use' in September and proceeded with public consultation. The newly disclosed approval cases totaled 2,186 from 2020 to the present, broken down as follows: 472 cases in 2020, 572 in 2021, 266 in 2022, 485 in 2023, 276 in 2024, and 115 this year. Meanwhile, a total of 377 rejection cases were disclosed over 13 years, from 2013 to 2025. Specifically, from 2020, when approval cases began to be disclosed, 208 cases were rejected. Examining the trend of rejections by year shows: 33 rejections in 2020, 86 in 2021, 38 in 2022, 44 in 2023, 6 in 2024, and 1 this year. This data indicates a significant reduction in rejection cases over the last two years. The disclosed rejection data for this year includes one additional rejection for Mabthera (rituximab). The application was filed citing expectations that the drug would be more cost-effective, have fewer side effects, and offer higher therapeutic efficacy than alternative drugs. Yet, it was denied because the submitted evidence did not substantiate the claims.