- LOGIN
- MemberShip
- 2026-06-10 19:52:15
- Opinion
- [Desk’s View] Unmet needs remain in immuno-oncology
- by Eo, Yun-Ho Apr 13, 2026 09:12am
- Immuno-oncology has now become quite a fairly common term. It is a term even ordinary people are likely to have heard at least once. More than a decade has already passed since the term was first introduced to Korea. At present, immuno-oncology drugs have expanded their indications across various cancer types and established themselves as a major pillar of cancer treatment. Whether the growing number of indications receive coverage has become an important gateway that determines treatment access.The extent to which the clinical value of new treatment options should be reflected during reimbursement review remains a persistent concern. For the government, it is a matter of striking a balance between the financial burden and the clinical benefits offered by new drugs.The upcoming Cancer Disease Deliberation Committee of the Health Insurance Review and Assessment Service is one place where this question constantly comes up for debate. At this month’s meeting, reimbursement for the ‘Opdivo (nivolumab)’ and ‘Yervoy (ipilimumab)’ combination as first-line treatment for hepatocellular carcinoma and non-small cell lung cancer will be presented for deliberation. At the meeting held last October, the combination was rejected for both liver and lung cancers.In hepatocellular carcinoma, following Tecentriq (atezolizumab) plus Avastin (bevacizumab), Imfinzi (durvalumab) plus Imjudo (tremelimumab) have also been added to the reimbursement list. In non-small cell lung cancer as well, an immuno-oncology-based treatment strategy has already taken hold, with ‘Keytruda (pembrolizumab)’ already reimbursed as monotherapy and combination therapy for 4 years.With immuno-oncology drug combinations already reimbursed, attention is now turning to whether reimbursement criteria will be set for the new Opdivo-Yervoy combination, as the addition must be more than simply another treatment option to pass review.In this regard, hepatocellular carcinoma remains a cancer type with frequent recurrence, poor prognosis, and high mortality rates, and many patients begin treatment with impaired liver function. Due to these disease characteristics, key evaluation criteria include whether the treatment option can provide deep and durable responses, long-term survival, and long-term survival benefit regardless of liver function status.The Opdivo and Yervoy combination is the treatment option that has demonstrated the longest survival data in first-line treatment for hepatocellular carcinoma. In clinical trials, it recorded a median overall survival (mOS) of 23.7 months, with a survival rate of 31% at 48 months. In addition, in an Asian patient subgroup analysis, a median overall survival (mOS) of 34.0 months, a 3-year survival rate of 49%, an objective response rate of 37%, and a complete response rate of 10% were reported. Compared with existing immuno-oncology combinations, whose mOS typically does not exceed 20 months, these are significant results.In particular, the Opdivo-Yervoy combination significantly reduced the risk of death by 25% versus the control arm, even in patients with impaired liver function classified as ALBI grade 2/3, demonstrating a degree of mortality risk reduction comparable to that seen in patients with preserved liver function.Its use in non-small cell lung cancer also warrants attention. Although Keytruda-based regimens have effectively become the cornerstone of first-line treatment, it is difficult to say that they fully address the treatment needs of all patient subgroups. In practice, there are still patient groups, such as those with PD-L1-negative tumors or squamous histology, for whom long-term survival benefit under existing immuno-oncology treatment settings has been reported only to a limited extent.As the Opdivo-Yervoy combination demonstrates consistent survival improvements in these patient groups, regardless of PD-L1 expression or histology, it has been discussed as a viable alternative.Ultimately, the core of the Cancer Disease Deliberation Committee review should not be on whether another option should be added to the reimbursement list. Real deliberation should be made on whether the current reimbursement system is offering a sufficient range of treatment choices in practice, and to what extent unmet treatment needs in specific patient groups should be reflected in deliberations. It would be difficult to accept a conclusion that the current system is sufficient merely because options already exist. When it comes to immuno-oncology drugs, unmet needs remain.
- Policy
- K-Bio Q1 sales hit record high…tops $2 billion
- by Lee, Tak-Sun Apr 13, 2026 09:12am
- Exports of Korean biopharmaceuticals in the first quarter of this year were at a record high. In particular, exports of biosimilars to Europe increased significantly.The Ministry of Food and Drug Safety (MFDS, Minister Yu-kyoung Oh) announced on the 10th that the export volume of South Korea's biopharmaceuticals in the first quarter of 2026 reached an unprecedented record of $2 billion (estimated), an 11.1% increase compared to the export value of the first quarter of last year.The MFDS explained that this is driven by an increase in the market share of K-biopharmaceuticals and by the expansion of competitiveness among biopharmaceutical Contract Development and Manufacturing Organizations (CDMOs).According to export value by year for the first quarter, it recorded $1.5 billion in 2024, followed by $1.8 billion in 2025 (a 20% increase from the previous year), and $2 billion this year (an 11.1% increase). Biopharmaceuticals accounted for 71% of the total pharmaceutical export value of $2.8 billion in the first quarter of this year."Export value by year for Q1": $1.5 billion in 2024, followed by $1.8 billion in 2025 (a 20% increase from the previous year), and $2 billion this year (an 11.1% increase). "Top 5 countries for exports in Q1": The country with the largest exported value in the first quarter of 2026 was Switzerland, recording $340 million (17.0% of total exports). Source: Korea Customs Service HS code and Korea Trade Statistics Promotion InstituteBy month, exports in January and February increased by 11.9% and 25.4% year-on-year to $660 million and $690 million, respectively. March exports were similar to the same period last year at $650 million, showing steady export figures from January through March.The country with the largest exported value in the first quarter of 2026 was Switzerland, recording $340 million (17.0% of total exports). The United States followed this at $330 million (16.5%) and Hungary at $300 million (15.0%). Exports to the top five countries accounted for 68.4% of the total.Exports to Switzerland increased by 70% (+$140 million) compared to the same period last year, rising from the 4th largest export destination in the first quarter of last year to 1st place this year.Exports to the United States decreased by $40 million (-12.6%) year-over-year, accounting for 16.5% of the first quarter export value. Exports to Hungary increased by $50 million (+20.2%) year-over-year.Analysis suggests that the increase in exports to Europe is due to a combination of cooperation with global pharmaceutical companies, technology exports, and a favorable environment for biosimilars.The MFDS explained that it is strengthening the global competitiveness of domestic biopharmaceuticals and helping them enter overseas markets by advancing rational regulatory innovation and providing customized information, as well as by actively pursuing regulatory diplomacy with major exporting countries.Aligning with the rapid growth of the biopharmaceutical CDMO market, the 'Special Act on Regulatory Support for Biopharmaceutical CDMO Companies' was enacted. By introducing a registration system for export manufacturing, an institutional foundation was established, enabling CDMO companies for export purposes to enter the global market without a pharmaceutical manufacturing license.Furthermore, the MFDS is advancing innovations in the biopharmaceutical approval and review process and in full-cycle regulatory support, enabling safe treatments to be launched faster than anywhere else in the world.To enable domestic biotech companies to enter the international market quickly, the MFDS has simplified the documents required for a preliminary GMP evaluation (from 11 to 4). It has preemptively promoted the 'Raw Material Manufacturing Site Certification Pilot Project' to support the entry of domestic biopharmaceutical raw materials into the global market.In addition, to respond to varying licensing systems and regulatory environments by country, the "Click! Global Biopharmaceutical Information Service" is operated to systematically provide regulatory information for 24 major countries, including the U.S., Europe, and Southeast Asia, along with the latest guidelines and translations to address local regulatory changes.An official from the MFDS stated, "We will continue to strengthen the international competitiveness of our biopharmaceuticals through rational regulatory improvement and institutional and technical support. We will also create an environment where the public can use them with safety management of biopharmaceuticals."

- Policy
- PVA discount for innovative companies to increase to 50%
- by Jung, Heung-Jun Apr 13, 2026 09:12am
- With the reduction rate for price cuts under the price-volume agreement (PVA) for innovative pharmaceutical companies raised to 50%, the outlook for pharmaceutical companies is expected to diverge depending on whether the existing conditions related to repeated cuts remain in place.This is because the number of beneficiaries would decrease significantly if an additional condition requiring 3 price cuts within 5 years were imposed. This is expected to be a major point of contention during discussions over detailed requirements.According to the industry and relevant institutions on the 10th, while the increase in the PVA reduction rate for innovative companies has been decided, the detailed conditions have not yet been finalized.AI-generated ImageIn March, the Health Insurance Policy Deliberation Committee approved strengthened post-listing management preferential treatment for innovative companies by increasing the reduction rate for PVA-driven price cuts. The plan involves raising the reduction rate for price cuts from 30% to 50% when price reductions occur due to increased usage volume.For example, if usage increases and the drug price reduction rate is set at 4%, the cut rate for innovative companies would be lowered to 2%.Under current guidelines for PVA negotiations, a condition is attached for drugs that have undergone repeated cuts. To qualify for the reduction, a drug must have reached an agreement in negotiations at least twice over a five-year period, and the manufacturer must be either an innovative pharmaceutical company or a company recognized by the Health Insurance Review and Assessment Service (HIRA) as having R&D expenses accounting for 10% or more of its revenue.If a drug currently under negotiation has received a third negotiation order within 5 years prior to the end of the analysis period, the pharmaceutical company may submit documents to receive a 30% reduction.The key issue is whether these additional conditions will also apply under the new 50% reduction rate scheme. If eligibility is limited to products whose usage has increased enough to warrant 3 rounds of negotiations, the number of eligible items will decrease significantly.According to the NHIS, 17 items received a 30% reduction in the negotiations 2 years ago because they had been subject to price reductions 3 or more times within 5 years.The industry is hopeful that the specific conditions may change, as the Health Insurance Policy Deliberation Committee approved the 50% increase in the reduction rate without specifying any concrete conditions.In particular, the industry maintains that a 50% reduction should be granted without additional conditions to incentivize innovative companies and R&D investment.An official from a domestic pharmaceutical company expressed concern, stating, “If this does not apply to the third round of negotiations, specific implementation methods must be determined, such as whether it applies only to the first round or whether reductions will also apply to the second and third rounds. However, limiting it to the third round would significantly reduce the number of eligible items.”
- Company
- Final Zemiglo use patent invalidated in Korea
- by Kim, Jin-Gu Apr 13, 2026 09:11am
- The dispute surrounding the use patent for LG Chem’s diabetes treatment Zemiglo (gemigliptin) has ended with a final victory for generic companies. With this ruling, generic companies will be able to launch generic versions of Zemiglo after the substance patent expires in January 2030.Supreme Court issues discontinuance of trial on LG Chem’s appeal… use patent finally invalidatedAccording to the industry sources on the 10th, the Supreme Court issued a discontinuance of trial in the final appeal of the Zemiglo use-patent invalidation case filed by LG Chem against Celltrion Pharm, Dongkoo Bio & Pharma, Daehwa Pharmaceuticals, Jeil Pharmaceutical, and Boryung.A discontinuance of trial means that the Supreme Court affirms a lower court’s ruling without reviewing the merits of the case, having determined that the grounds for the appeal do not meet legal requirements. Consequently, the second-instance ruling, in which LG Chem lost, has been finalized. The use patent for Zemiglo has therefore been invalidated.LG Chem and generic drug companies had been in dispute over the use patent, which expires in October 2039. This patent covers the combined administration of gemigliptin and insulin. Celltrion Pharm and others filed a petition for invalidation in 2023, arguing that the patent lacked inventive step.The Intellectual Property Trial and Appeal Board (first instance) and the Intellectual Property Court (second instance) both ruled in favor of the generic companies. The Supreme Court then reached the same conclusion, putting an end to a legal battle that had lasted nearly 3 years.Impact of the final invalidation ruling… scope-confirmation litigation previously won by LG Chem also heading toward closureThis ruling is expected to influence a separate litigation regarding the scope of rights currently underway concerning the same use patent.Until now, disputes over Zemiglo’s use patent had proceeded along two separate tracks - the ‘invalidity lawsuit’ and the ‘scope of rights confirmation lawsuit.’ While the generic company won both disputes in the first instance, the rulings diverged in the second instance. While the generic drug companies prevailed in the invalidity suit regarding the use patent, the original manufacturer, LG Chem, won the dispute over the scope of rights.Because of those conflicting second-instance rulings, uncertainty grew over the timing of the early generic launch. At the time, there were concerns that if LG Chem ultimately succeeded in defending the patent, a generic launch could be delayed until after 2039.However, the situation has now reversed with the Supreme Court ruling. Legally, once a patent is definitively invalidated, the rights associated with it are deemed to have never existed from the outset. That means the favorable ruling LG Chem obtained in the scope-confirmation litigation loses legal effect, because the patent in question, which served as the basis for comparison, is now interpreted as “non-existent.”From the perspective of generic drug companies, this ruling effectively allows them to bring forward the launch of Zemiglo generics by 9 years, to a date after January 2030, when the substance patent expires. Although Zemiglo has a salt and hydrate patent set to expire in October 2031, generic drug companies have already successfully circumvented it.Zemiglo is LG Chem’s flagship drug. According to market research firm UBIST, the combined prescription sales of the ‘Zemiglo family’, which includes Zemiglo, Zemimet, Zemidapa, and Zemiro, totaled KRW 159.1 billion last year, a 4% increase from the previous year. Among these, Zemiglo alone recorded KRW 41.4 billion in prescription sales, accounting for 26% of the total family product prescriptions.
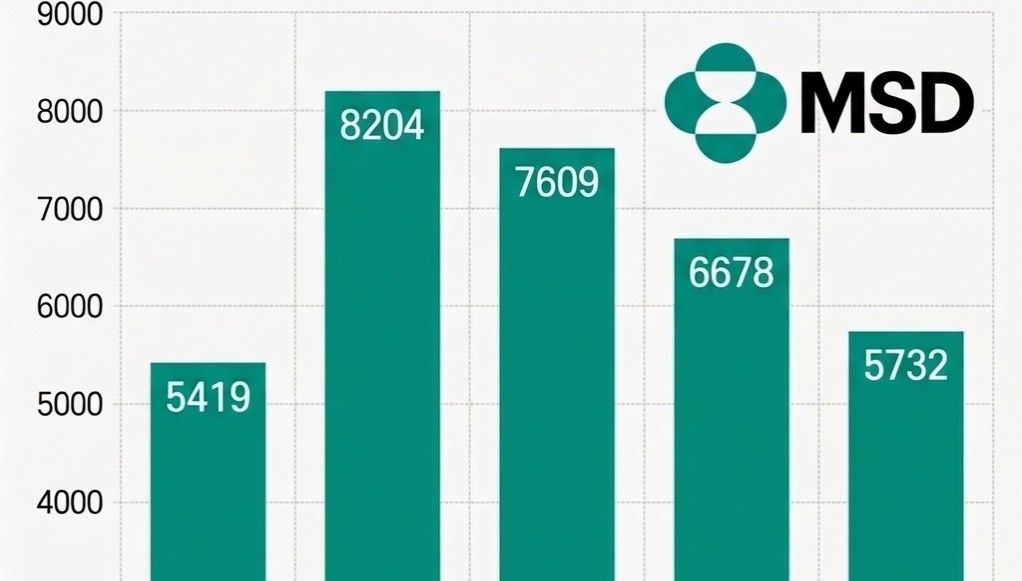
- Company
- MSD Korea sales 30%↓ in three years
- by Son, Hyung Min Apr 13, 2026 09:11am
- MSD Korea's performance continued to decline due to lower demand for COVID-19 treatments. As sales from treatments that drove performance during the pandemic have rapidly shrunk, existing core products have failed to offset the loss.According to the Financial Supervisory Service's electronic disclosure system on the 13th, MSD Korea's sales decreased by 14.2%, from KRW 667.8 billion in 2024 to KRW 573.2 billion last year. During the same period, operating profit dropped 13.0%, from KRW 24.9 billion to KRW 21.6 billion.MSD Korea Sales Trend by Year (unit: KRW 100 million). MSD Korea's sales decreased by 14.2%, from KRW 667.8 billion in 2024 to KRW 573.2 billion in 2025.The primary reason for the sales decline is the lack of supply of the COVID-19 treatment 'Lagevrio (molnupiravir).' MSD Korea explained that the absence of a supply contract with the Korea Disease Control and Prevention Agency (KDCA) last year affected the sales decrease.In fact, MSD Korea's sales have shown significant volatility, reflecting demand for COVID-19 treatments. Revenue peaked at KRW 820.4 billion in 2022 when demand was the highest, but subsequently decreased to KRW 760.9 billion in 2023 and KRW 667.8 billion in 2024 following the transition to the endemic phase. Compared with last year's sales of KRW 573.2 billion, revenue has shrunk by 30.1% over the past three years.Despite having a strong lineup of major products, including the immunotherapy 'Keytruda (pembrolizumab),' the cervical cancer vaccine 'Gardasil,' and the pneumococcal vaccines 'Vaxneuvance' and 'Capvaxive,' it was not enough to fill the void left by the end of the COVID-19 sales.However, the company continued investing in its Research and Development (R&D).According to data released by MSD Korea, the company invested 78 billion KRW in R&D last year, accounting for approximately 14% of its revenue, and has consistently invested over KRW 70 billion annually for the past five years. Despite the short-term performance decline, the company appears to be continuing its strategic investments to secure a foundation for medium- to long-term growth.New Indications·Pipeline Additions…Seeking a Rebound in PerformanceWhile performance has declined due to the sales void from COVID-19 treatments, MSD Korea is seeking a rebound opportunity by reorganizing its portfolio around oncology, vaccines, and rare diseases.The scope of Keytruda was rapidly expanded this year, with 11 additional indications, including triple-negative breast cancer and endometrial cancer, added to the reimbursement list. Furthermore, reimbursement for combination therapy with 'Padcev (enfortumab vedotin)' in urothelial carcinoma is also imminent.RSV preventive antibody injection 'Enfloncia (clesrovimab)' Keytruda has become a pillar of treatment with expanded reimbursement scope as a standard of care (SOC) across major solid tumors. This treatment has the most indications among drugs authorized in Korea.At the same time, efforts to develop new growth engines for infectious diseases are underway. MSD Korea has applied for the authorization of 'Enfloncia (clesrovimab),' an RSV preventive antibody injection for neonates and infants, and there is talk of possible approval in the second half of this year.Enfloncia is a long-acting monoclonal antibody that, in Phase 2b/3 clinical studies, demonstrated reductions of 60.5% in the occurrence of RSV-related lower respiratory tract infections and 84.3% in the risk of hospitalization.In addition, the reimbursement process for 'Winrevair (sotatercept),' a treatment for pulmonary arterial hypertension (PAH), is accelerating following its inclusion in the pilot project for concurrent authorization, evaluation, and negotiation.Winrevair is the first approved activin signaling inhibitor (ASI) in pulmonary arterial hypertension and offers a new mechanism of action after 20 years. This treatment works by blocking excessive activin signaling. This protein complex promotes cell proliferation in pulmonary arterial vessels, and restores the balance with anti-proliferation signals to induce reverse remodeling, normalizing altered vascular structures.As the impact from the termination of the COVID-19 special demand is being reflected, the expanded reimbursement for major products and the introduction of new drugs are expected to be key drivers of a future performance rebound.
- Policy
- Label updated for hypertension drug nebivolol
- by Lee, Tak-Sun Apr 10, 2026 08:27am
- Nebivolol original ‘Nebilet’The label for the antihypertensive drug nebivolol will now include the statement, “Beta-blockers may further increase the risk of severe hypoglycemia when co-administered with sulfonylureas (SU).”This follows safety measures taken by the European Medicines Agency (EMA), with Korea’s Ministry of Food and Drug Safety (MFDS) moving to revise product labeling.While concerns about hypoglycemia due to symptom masking when beta-blockers are used in combination with sulfonylureas (SU) are well known, the latest change is intended to further emphasize the severity of the risk and strengthen monitoring.On the 6th, MFDS announced draft labeling revisions for nebivolol-containing products and will collect feedback through the 21st.This draft is based on safety information from the European Medicines Agency (EMA). Nebivolol is a third-generation beta-blocker.According to the proposed amendment, the following statement will be added to the General Precautions section: “Beta-blockers may further increase the risk of severe hypoglycemia when co-administered with sulfonylureas. Patients with diabetes should be advised to carefully monitor their blood glucose levels.”Additionally, the following statement will be added to the “Drug Interactions” section. “Concomitant use of beta-blockers and sulfonylureas may increase the risk of severe hypoglycemia.”Current labeling already advises caution when administering the drug to patients with diabetes. This is because beta-blockers, including nebivolol, can mask specific symptoms of hypoglycemia, such as tachycardia and palpitations,As a result, patients may fail to recognize hypoglycemia and progress to severe hypoglycemia.Consequently, this may increase the likelihood that patients will progress to severe hypoglycemia without recognizing the symptoms.The Interactions section currently also includes a statement that co-administration with insulin or oral antidiabetic agents may mask specific symptoms of hypoglycemia (palpitations, tachycardia).However, the current labeling does not specifically warn against co-administration with sulfonylureas (SU) among antidiabetic agents.Last year, the EMA reviewed evidence suggesting that the risk of severe hypoglycemia may increase when nebivolol is used in combination with sulfonylureas and required that this information be added to the product label.MFDS appears to have prepared the Korean revision as a follow-up measure to the EMA’s actions.Currently, there are 25 nebirol-containing products approved in Korea. These include the original “Nebilet Tab” (Menarini Korea), the low-dose “Nebilet M Tab” (Kwangdong Pharmaceutical), and the nebirol-rosuvastatin combination drugs “Nebirosta Tab” (Elyson Pharm) and “Crebista Tab” (Arlico Pharmaceutical).According to UBIST, Nebilet recorded KRW 8.6 billion in outpatient prescriptions last year.

- Policy
- Opposition to labeling obesity drugs as 'misuse drug'
- by Lee, Tak-Sun Apr 10, 2026 08:27am
- AI-generated imageVoices are growing against the plan to designate GLP-1 obesity treatments, such as Wegovy and Mounjaro, as "high potential misuse and abuse." Critics point out that such a designation could instead drive patients toward illegal distribution channels.Analysis suggests that other countries are already putting effort into establishing an environment for safe use by monitoring distribution and prescription, rather than regulating the drug itself.On the 8th, the Ministry of Food and Drug Safety (MFDS) will discuss whether to designate these treatments through the Central Pharmaceutical Affairs Advisory Committee (CPAC).Currently, this category includes erectile dysfunction treatments, premature ejaculation treatments, and anabolic steroids. Once designated, the phrase "high potential misuse and abuse" must be displayed on the product packaging, and sales without a prescription are prohibited even in areas exempt from the separation of prescribing and dispensing.The MFDS's push for this designation is due to issues such as non-face-to-face prescribing, online black-market transactions, and indiscriminate off-label use, following the immense popularity of Wegovy and Mounjaro. Following discussions in last year's parliamentary audit, health authorities have been reviewing this designation.However, the industry is voicing concern that the designation could lead to illegal distribution, resulting in a balloon effect.However, the industry warns that labeling these drugs as potentially misused could shrink the patient base and prevent those who genuinely need treatment from receiving it.They argue that global trends focus on regulating the distribution and prescription stages rather than the drugs themselves.In Japan, as the prescribing of Ozempic (semaglutide) for obesity purposes increased, guidelines were established for prescribing institutions. According to an analysis conducted by the University of Tokyo research team of Japanese medical institution websites, institutions that advertised non-reimbursed prescriptions for GLP-1 receptor agonists exhibited significantly lower information quality. Furthermore, a large majority of these institutions were found to violate pharmaceutical advertising guidelines.Japan's Ministry of Health, Labor and Welfare (MHLW), when approving health insurance coverage for Wegovy in February 2024, set strict standards for prescribing institutions and patient lifestyle habits.Australia is also focusing on blocking unofficial distribution channels. From October 2024, Australia's Therapeutic Goods Administration (TGA) excluded GLP-1 drugs from the pharmacy compounding exemption. The system is designed to allow pharmacies to prepare similar drugs in response to drug shortages or to provide customized preparation for a specific patient.They also intensified advertising regulations, imposing fines of approximately 198,000 AUD (about 200 million KRW) for illegal advertisements on telehealth platforms.Japan and Australia have clear medical evidences and are focusing on monitoring illegal distribution and prescriptions, rather than designating obesity drugs that are approved by the regulatory authority as having the potential to be misused and abused.The WHO also emphasized 'regulated distribution networks, prescriptions by qualified medical professionals, and strong supervision' in its December 2023 global guidelines to counter the spread of counterfeit and substandard GLP-1 products.An industry official stated, "Major countries are setting standards for eligible prescribing institutions and restrictions on non-face-to-face prescriptions," adding, "They are putting effort into monitoring whether prescriptions align with BMI and comorbidities." The official added, "The Korean government and medical community should develop systematic management plans that block cosmetic use while protecting patient access for those who truly need treatment."There is also concern that the negative stigma of being a "high potential to misuse and abuse" could affect severely obese and type 2 diabetic patients. Another industry official stated, "Designation of 'high potential to misuse and abuse' mandates labeling, and the label points at all obesity patients. Not only those with severe obesity but also type 2 diabetic patients would receive medication stamped with 'potential to misuse'."Furthermore, there is a risk that patients who find it harder to obtain legitimate prescriptions may turn to unverified online purchases or illegal distribution channels. With cases of fraud and harm from illegal transactions already present in Korean online communities, critics fear that the official designation could expand this illegal market.
- Policy
- Samsung Bioepis-Hanmi joint sales 'Obodence' wins nod for IIT
- by Lee, Tak-Sun Apr 10, 2026 08:27am
- Product photo of ObodenceA large-scale investigator-initiated trial (IIT) for 'Obodence,' Samsung Bioepis' denosumab biosimilar, will begin.Led by Yeouido St. Mary's Hospital, this clinical trial is drawing significant attention as a large-scale study involving 13 medical institutions across South Korea. It is expected that this trial will provide clearer evidence of Obodence's efficacy, specifically in Korean patients.On the 8th, the Ministry of Food and Drug Safety (MFDS) approved the clinical trial protocol for 'Obodence Prefilled Syringe' requested by Catholic University of Korea Yeouido St. Mary's Hospital.The clinical trial aims to evaluate the efficacy of the denosumab biosimilar in postmenopausal women diagnosed with advanced osteopenia. It is designed as a multicenter, randomized, open-label study, corresponding to a Phase 4 post-marketing clinical trial.Obodence was launched in July of last year as a biosimilar to Prolia, an osteoporosis treatment. Prolia has dominated the osteoporosis market in South Korea, recording annual sales of approximately KRW 180 billion, given its strong efficacy and the convenience of once-every-six-month administration.The biosimilar market took off last year. In March, the first biosimilar, Celltrion's 'Stoboclo,' began joint sales with Daewoong Pharmaceutical. In July, Samsung Bioepis launched Obodence in the market through a partnership with Hanmi Pharmaceutical.The market share for biosimilars is on the rise, with Stoboclo recording sales of KRW 11.8 billion within its first 10 months.Samsung Bioepis, with Obodence launched slightly later than Stoboclo, is now focusing on marketing strategies to secure trust through clinical evidence.Notably, Samsung Bioepis is emphasizing to medical professionals that Obodence demonstrated results highly similar to the original in Phase 3 trials that included Korean participants. The Phase 3 study involved 457 postmenopausal patients with osteoporosis across five countries, including South Korea.In this regard, the current IIT is expected to serve as an opportunity to accumulate further evidence of its efficacy in Koreans. Given that the trial is being conducted across 13 domestic hospitals, it is anticipated to include a larger number of Korean subjects than the Phase 3 trial did.However, the company clarified that, as this is an investigator-initiated trial, it does not necessarily reflect the firm's specific corporate objectives.Stoboclo also received approval for an IIT last February. The study, conducted by Ajou University Hospital, is designed to evaluate the efficacy of denosumab in obese subjects treated with GLP-1 receptor agonists who exhibit weight loss and bone metabolism risk factors.
- Company
- AZ launches Tezspire in Korea with expanded indication
- by Son, Hyung Min Apr 10, 2026 08:27am
- AstraZeneca Korea (CEO Eldana Sauran) announced on the 8th the domestic launch of Tezspire (tezepelumab) as an add-on maintenance treatment for severe asthma and chronic rhinosinusitis with nasal polyps (CRSwNP).With this domestic launch, Tezspire has simultaneously expanded its indication to include its use as an add-on maintenance treatment for adults with inadequately controlled CRSwNP, broadening its use as an anti-TSLP (Thymic Stromal Lymphopoietin) treatment option for severe asthma to CRSwNP.TSLP is a driver of multiple inflammatory responses and is expressed at higher levels in CRSwNP patients than in patients without polyps. Tezspire is an anti-TSLP monoclonal antibody that blocks TSLP activity at the upstream level of inflammatory pathways. Tezspire’s clinical efficacy and safety profile were confirmed in the global Phase III WAYPOINT trial.The WAYPOINT study was a multicenter, randomized, double-blind, placebo-controlled Phase III clinical trial conducted in 10 countries involving 408 patients aged 18 years and older with CRSwNP who had severe, uncontrolled symptoms.Results showed that at Week 52, the Tezspire treatment group demonstrated statistically significant improvements compared to the placebo group, with a decrease of -2.07 in the Nasal Polyps Score (NPS), which assesses the size and extent of nasal polyps, and a decrease of -1.03 in the Nasal Congestion Score (NCS), which assesses the degree of nasal congestion. Furthermore, these improvements were observed as early as week 4 and week 2 of treatment, respectively, and were sustained through week 52.Ji-young Kim, Executive Director of AstraZeneca Korea’s Respiratory Division, said, “We are pleased that we were able to expand Tezspire’s indication in Korea following FDA approval in October last year for CRSwNP. Clinical trials confirm Tezspire can be an effective treatment option not only for asthma but also for patients with CRSwNP, and we expect it to help patients manage their respiratory conditions.”Rhinosinusitis is characterized by two or more symptoms, including nasal congestion, nasal obstruction, or a runny nose, and becomes chronic when these symptoms persist for 12 weeks or longer. Additionally, when accompanied by nasal polyps, it is classified as CRSwNP.
- Company
- Boston Scientific Korea’s sales surpass ₩200 billion
- by Hwang, byoung woo Apr 10, 2026 08:26am
- Boston Scientific Korea has surpassed KRW 200 billion in sales following portfolio restructuring.Analysts attribute this revenue expansion to the robust growth of the Pulse Field Ablation (PFA) system, coupled with concurrent growth across all major therapeutic areas.According to a recent audit report, Boston Scientific Korea posted KRW 218.5 billion in sales in 2025, up 18.4% from KRW 184.6 billion the previous year.This marks the first time the company has surpassed KRW 200 billion in revenue since entering the Korean market, continuing its four-year growth streak from KRW 151.6 billion in 2022, KRW 175.3 billion in 2023, and KRW 184.6 billion in 2024.Operating profit also grew in tandem with topline growth, from KRW 9 billion in 2022, KRW 10.5 billion in 2023, KRW 11 billion in 2024, to KRW 13 billion in 2025.Increased PFA procedures for arrhythmia drive growthThe key driver behind the company’s growth lies in changes to the business portfolio. The product lines currently supplied by the company to domestic medical institutions span the cardiovascular, oncology, and urology fields. After previously attempting to enter the structural heart disease segment with TAVI before discontinuing the business, the company restructured its portfolio.Representative products include the AVIGO Plus coronary ultrasound imaging device, the TheraSphere liver tumor embolization device, and the Rezum system for benign prostatic hyperplasia.Among these various products, the PFA system had the greatest impact on growth last year.Unlike conventional radiofrequency ablation or cryoballoon ablation, PFA selectively destroys myocardial cells only, reducing procedure time by more than half and lowering complication risks, thereby expanding its influence in major domestic general hospitals.The Korean PFA market is currently contested by Boston Scientific, Medtronic, and Johnson & Johnson (J&J).Boston Scientific was the fastest to enter the Korean market, securing catheter certification for its FARAPULSE platform in April 2024 and generator approval in September.Analysts attribute this growth to the expanding market for electrophysiological procedures, driven by an increase in atrial fibrillation patients amid an aging population, with a recurring revenue model centered on related catheters and systems serving as the foundation for growth.In a Korea Health Industry Development Institute report on PFA, Professor Bo-young Joung of the Department of Cardiology at Severance Hospital stated, “PFA cuts procedure time by half compared with conventional methods and lowers complication risk, leading to high satisfaction among both physicians and patients. Currently, 35% of atrial fibrillation ablation procedures at Severance are performed using PFA, and this trend is expected to continue.”Given this, Boston Scientific Korea’s growth momentum is expected to strengthen further.Rezūm expansion and global M&A broaden business scopeAnother factor contributing to growth is the continued expansion of the Rezūm System, a medical device introduced in 2023 for benign prostatic hyperplasia, which has now surpassed 6,000 cumulative procedures in Korea.The Rezūm System received approval from the U.S. Food and Drug Administration (FDA) in 2015, obtained authorization from the Ministry of Food and Drug Safety in 2022, and was designated as a new health technology by the Ministry of Health and Welfare in 2023. Currently, Rezūm procedures are expanding their scope of application in clinical settings as a treatment option that can be considered even for patients unsuitable for medication or surgery.Regarding this, Boston Scientific Korea Country Manager Ae Ri Jung said, “Achieving 6,000 Rezūm procedures is a meaningful milestone demonstrating its establishment as a treatment option for BPH in Korea. We will continue contributing to expanding treatment options that improve patients’ quality of life.”As the portfolio expands, the company appears to be strengthening its field sales capabilities. Looking at the sales and administrative expenses, promotional expenses rose from KRW 6.3 billion in 2024 to KRW 7.1 billion in 2025.In particular, as Boston Scientific is pursuing mergers and acquisitions (M&A) on a global scale involving tens of trillions of won, its influence in the domestic medical market is expected to continue to grow in the future.The company acquired Axonics in 2024 to strengthen its urology portfolio, and in January, decided to acquire neurovascular treatment company Penumbra.In addition, it has also recently fully integrated Valencia Technologies, a urinary incontinence treatment company, rapidly expanding its portfolio.As the company expands its business scope to include new disease areas in addition to those that generate synergies with existing businesses, its business scale in the Korean market is also expected to grow.