- LOGIN
- MemberShip
- 2026-06-10 19:52:15

- Company
- "Data analysis of 500,000 patients…clinical utility of choline alfoserate reconfirmed"
- by Kim, Jin-Gu May 07, 2026 10:29am
- 'Choline alfoserate expert forum' recently hosted by DailyPharmA large-scale real-world data (RWD) analysis of 500,000 Korean patients with mild cognitive impairment (MCI) has reconfirmed the clinical utility of choline alfoserate in reducing the risk of progression to dementia.Experts evaluate that this study significantly strengthens the evidence base for prescribing choline alfoserate. It is considered academically meaningful because it revalidated the efficacy evidence from existing randomized controlled trials (RCTs) using massive datasets from actual clinical settings.Analysis of 500,000 Korean MCI patients...“Reduction in risks for both Alzheimer’s disease‧vascular dementia”At the 'Choline alfoserate expert forum' recently hosted by DailyPharm, Professors Han-kyeol Kim (Wonju Severance Christian Hospital), Choi Hojin (Hanyang University Guri Hospital), Lee Chan Nyung (Korea University Anam Hospital), and Kim Geon Ha (Ewha Womans University Mokdong Hospital), all neurologists, focused on the results of the study titled 'Effect of Choline Alfoserate: NHIS Cohort Study,' published last year.Professor Han-kyeol Kim (Wonju Severance Christian Hospital)Professor Han-kyeol Kim, who delivered the keynote presentation, introduced the research findings based on the National Health Insurance Service (NHIS) big data, which followed 508,107 patients newly diagnosed with MCI between 2013 and 2016.The study results showed that the choline alfoserate user group had a significantly lower risk of progression to Alzheimer’s dementia by 10.1% (HR 0.899) and vascular dementia by 16.8% (HR 0.832) compared to the non-user group. Furthermore, the risk of ischemic stroke decreased by 16.7% (HR 0.833), and the risk of hemorrhagic stroke decreased by 15.3% (HR 0.847).Professor Han-kyeol Kim said, "This was not a simple statistics of prescriptions. We linked health examination data to precisely adjust for variables such as smoking, alcohol consumption, income, and chronic diseases (hypertension, diabetes, dyslipidemia), as well as the duration of drug exposure to enhance objectivity," and added, "The study confirms that the use of choline alfoserate can be effective in reducing the risk of dementia progression and stroke in MCI patients, serving as a useful option for early intervention."Combining the scientific achievement of RCTs with the confirmed RWD… “Completing complementary evidence for dementia suppression”A discussion continued, chaired by Professor Choi Hojin, with Professors Lee Chan Nyung and Kim Geon Ha in attendance. They noted that this RWD study addresses the structural limitations of previous RCTs.The previous 'ASCOMALVA' study was designed as a multicenter, randomized, double-blind, controlled trial (RCT). The study targeted 210 patients with Alzheimer’s disease dementia accompanied by ischemic stroke, comparing a donepezil monotherapy group with a choline alfoserate + donepezil combination therapy group. The results confirmed significant improvements in the combination group across ▲the Mini-Mental State Examination (MMSE) ▲ Alzheimer's Disease Assessment Scale-Cognitive Subscale (ADAS-cog) ▲Instrumental Activities of Daily Living (IADL). Long-term follow-up over four years also showed that the rate of cognitive decline was inhibited in the combination therapy group.Experts agreed that this RWD study supplements the limitations of the RCT. Professor Choi Hojin evaluated, "The evidence for the efficacy of choline alfoserate has become even more robust by adding the analysis results of a large-scale Korean patient population to the ASCOMALVA findings," and added, "Results tracking the effects of drugs prescribed in the real world over a long period are highly meaningful as evidence from actual clinical practice, distinct from RCTs conducted in controlled environments."Professors Choi Hojin (Hanyang University Guri Hospital) ·Lee Chan Nyung (Korea University Anam Hospital) ·Kim Geon Ha (Ewha Womans University Mokdong Hospital)Professor Lee Chan Nyung stated, "While RCTs are suitable for proving drug efficacy in standardized environments, it is not easy to confirm actual conversion rates in slowly progressing diseases like dementia due to limitations in sample size and follow-up duration," and "This RWD study serves as powerful complementary evidence to existing RCTs by reaffirming the outcome of 'inhibiting dementia progression' through data accumulated over several years in actual clinical settings."“MFDS policy to enhance RWD reliability…increasing potential for utilization in clinical practice”Experts also emphasized that this study aligns with the recent government stance on the importance of RWD. In this regard, the Ministry of Food and Drug Safety (MFDS) announced the 'Guidelines for the Utilization of RWD/RWE in the Approval and Licensing of Medicines, etc.' in June 2024. Through this, the ministry presented specific criteria for actively recognizing RWD as clinical evidence for indication expansion or efficacy validation. In contrast, it had previously been used only as a supplementary tool for post-marketing surveillance (PMS) of side effects.The view among experts is that RWD research, which precisely analyzes NHIS big data from over 500,000 citizens, can yield new clinical evidence amid these changing regulatory environments. Professor Lee Chan Nyung emphasized, "As the vast RWD figures of 500,000 show, the dementia suppression effect confirmed in a large-scale patient group will serve as crucial evidence that gives clinical specialists the confidence to prescribe."Professor Kim Geon Ha said, "The 16.8% lower risk of progression to vascular dementia shown in this study holds great significance for the Korean elderly population with high vascular risk factors such as hypertension," and Kim concluded, "It raises the level of evidence for preemptive treatment of MCI patients in actual clinical settings."Professor Choi Hojin highlighted the public health significance of early intervention at the MCI stage. Choi added, "Delaying the onset of dementia by even just a few years can drastically reduce national medical costs and the caregiving burden on patients' families. Preemptive response through proven options will be a practical solution to alleviate the dementia burden in an aging society."
- Policy
- MFDS to revise labeling for 951 metformin products
- by Lee, Tak-Sun May 07, 2026 10:29am
- AI-generated imageThe government is pushing to revise the labeling for 951 metformin-containing products, the most widely used diabetes treatment in the market. This measure aims to explicitly state the potentially fatal side effects that may occur when metformin-containing products are administered to patients with hereditary mitochondrial disorders.According to industry sources on the 5th, the Ministry of Food and Drug Safety (Division of Drug Safety Evaluation) recently prepared a proposal to amend the approval conditions based on the European Medicines Agency (EMA)’s safety review regarding metformin-containing products and has begun collecting industry feedback.The key change is to restrict use and require immediate discontinuation in patients suspected or diagnosed with mitochondrial disorders.Under the proposed revision, metformin is not recommended for patients with ▲ MELAS syndrome (mitochondrial encephalopathy with lactic acidosis and stroke-like episodes) and ▲ MIDD (maternally inherited diabetes and deafness), due to increased risks of lactic acidosis and neurological complications.Additionally, if symptoms suggestive of MELAS or MIDD occur after administration, treatment should be immediately discontinued, and prompt diagnostic evaluation should be performed. The analysis indicates that metformin can affect intracellular mitochondrial metabolism, potentially causing severe energy metabolism disorders in patients with underlying genetic diseasesThe revision applies to a total of 951 products, including both monotherapy and combination drugs. As metformin is a core component of the domestic diabetes treatment market, most major pharmaceutical companies, including Chong Kun Dang, Daewoong, Hanmi, and Yuhan, are expected to be affected.The MFDS will accept feedback from relevant associations and companies until May 15. It will then comprehensively review the submitted comments and issue a final order to amend the approved product information.An MFDS official stated, “This measure aims to enhance the safe use of medicines for patients in Korea based on the latest safety information from the EMA. We ask that relevant associations and their member companies submit their review comments within the deadline.”
- Company
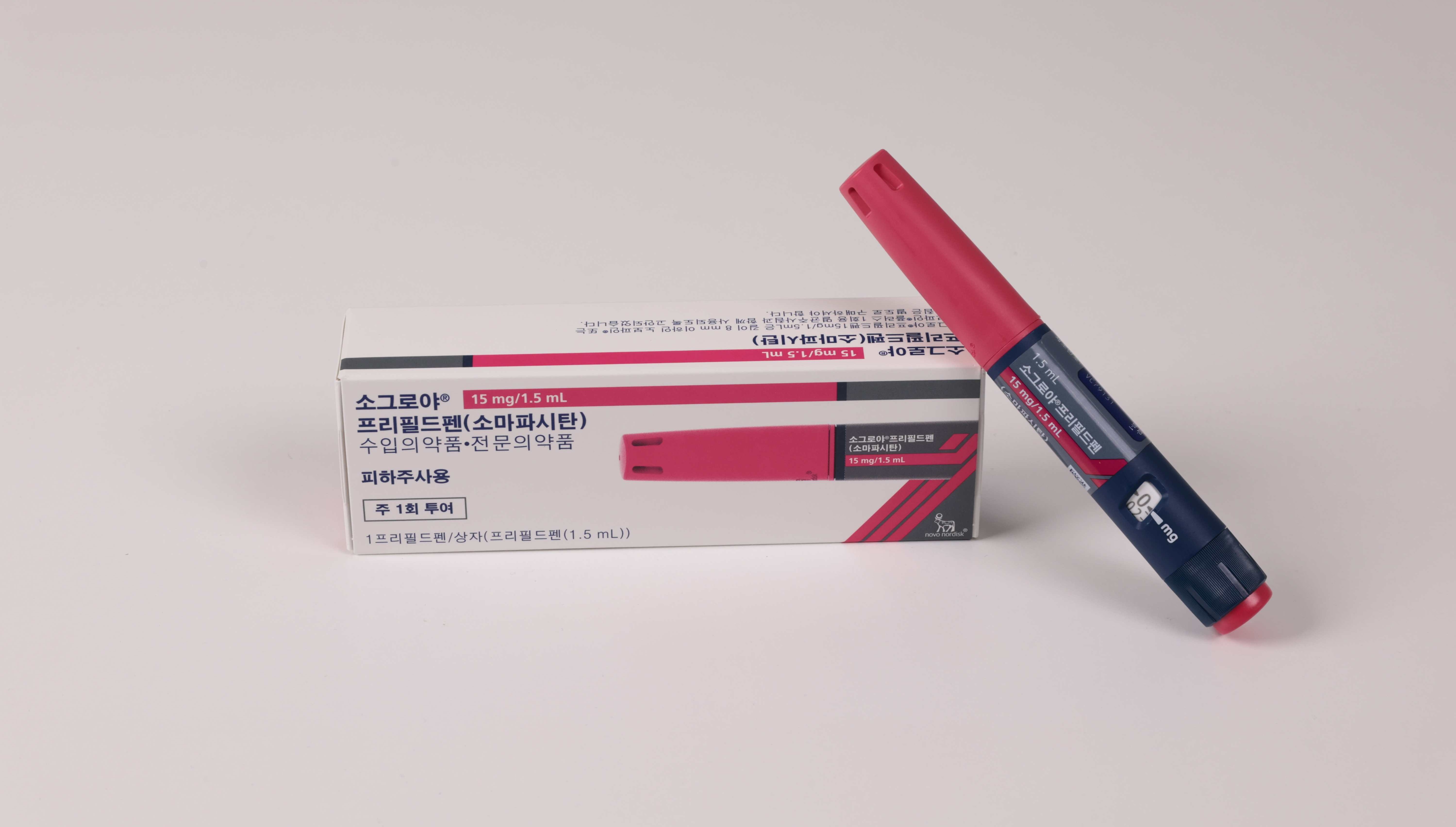
- Growth hormone deficiency drug Sogroya gains reimb in KOR
- by Son, Hyung Min May 07, 2026 10:29am
- Novo Nordisk Korea (General Manager: Kasper Roseeuw Poulsen) announced that starting on the 1st of this month, its long-acting, once-weekly growth hormone deficiency treatment ‘Sogroya Prefilled Pen (somapacitan)’ has been granted reimbursement for patients with growth hormone deficiency.Under the new reimbursement criteria, pediatric patients with growth hormone deficiency are eligible for Sogroya if they meet the following conditions: ▲ height below the 3rd percentile for chronological age, ▲ confirmed diagnosis through at least two growth hormone stimulation tests, ▲ and delayed bone age relative to chronological age.The recommended dosage is 0.16 mg/kg per week. Treatment is administered from a chronological age of 3 years until epiphyseal closure. However, reimbursement is limited to patients whose bone age falls within the range of 14–15 years for females and 15–16 years for males. However, patients within this category whose current height exceeds 153 cm (female) or 165 cm (male) must bear the full cost. In addition to children, Sogroya is covered for adult patients with growth hormone deficiency, provided certain criteria are met.Growth hormone deficiency is a condition characterized by delayed growth and may be accompanied by deficiencies in other pituitary hormones. Since consistent treatment is required until the end of the growth period, treatment adherence plays a critical role in treatment outcomes.The clinical efficacy and safety of Sogroya were confirmed in the global Phase III REAL4 trial.The REAL4 study was a randomized, parallel-group, open-label, active-controlled Phase III trial involving 200 treatment-naïve prepubertal pediatric patients with growth hormone deficiency. It evaluated the efficacy and safety of once-weekly Sogroya compared to once-daily growth hormone therapy.The primary endpoint was annualized height velocity (HV; cm/year) at Week 52. Secondary endpoints included changes in height velocity SDS, height SDS, the ratio of bone age (BA) to chronological age (CA), and IGF-1 SDS from baseline to Week 52.In the REAL4 study, Sogroya demonstrated non-inferiority in annual height velocity compared to daily growth hormone. At Week 52, the annual height velocity was 11.2 cm/year in the Sogroya group and 11.7 cm/year in the daily growth hormone group, showing comparable results.In terms of safety, the two treatment groups showed generally similar profiles. Most adverse reactions were mild or moderate in severity, and injection site reactions were reported in 5.3% and 5.9% of the Sogroya and daily growth hormone groups, respectively.Kasper Roseeuw Poulsen, General Manager of Novo Nordisk Korea, stated, “The reimbursement of Sogroya marks an important turning point in improving treatment access for patients with growth hormone deficiency. We expect that the once-weekly dosing option will improve treatment adherence, reduce the burden on patients and caregivers, and contribute to treatment continuity and improved quality of life.”

- Company
- Thyroid eye disease medicine 'Tepezza' lands in Korea
- by Son, Hyung Min May 07, 2026 10:28am
- Targeted therapy for thyroid eye disease (TED) 'Tepezza'Changes in treatment strategies are expected as the first targeted therapy for thyroid eye disease (TED) enters the Korean market.With follow-up candidates still in development or facing clinical hurdles with certain mechanisms, the possibility has been suggested that the market will remain centered on Amgen's already-commercialized Tepezza (teprotumumab).According to industry sources on the 7th, Amgen Korea recently obtained marketing authorization from the Ministry of Food and Drug Safety (MFDS) for Tepezza. The approved indication is for the treatment of adult patients with moderate-to-severe TED.TED is a rare disease in which autoimmune reactions cause inflammation and swelling of the orbital tissues. It causes proptosis, double vision, eye pain, and vision loss. If the disease progresses to a severe stage, it can lead to permanent disfigurement or even the risk of blindness.Until now, treatment in Korea has centered on symptomatic therapies such as steroids, radiation therapy, and orbital decompression surgery. However, critics have consistently pointed out that these have limitations in restoring structural changes such as proptosis or double vision.Tepezza is a monoclonal antibody that targets the Insulin-like Growth Factor-1 Receptor (IGF-1R), a key pathogenic mechanism in TED. It differentiates itself from existing treatments by targeting the disease to suppress the inflammatory response and tissue expansion.In the global Phase 3 OPTIC study, the proptosis response rate (a reduction of 2 mm or more) at 24 weeks was 83% for the Tepezza group and 10% for the placebo group. The diplopia (double vision) improvement rate also recorded 68% and 29%, respectively.In the OPTIC-J study of Japanese patients, the proptosis response rate was 89% in the Tepezza group and 11% in the placebo group.Tepezza already obtained regulatory agency authorization in the U.S. in 2020 and is currently the only approved targeted therapy for TED.FcRn inhibitors face hurdles… Delayed follow-up competitionSubcutaneous (SC) formulation 'Vyvgart (efgartigimod)'The competitive landscape for TED-targeted therapies remains limited, even in the global market.In particular, new drug candidates in the FcRn inhibitor class, which targeted to enter the TED market by reducing autoantibodies, are facing successive clinical barriers.Inhibition of the neonatal Fc receptor (FcRn), the protection receptor for Immunoglobulin G (IgG), is an approach that blocks the recycling pathway of IgG antibodies, thereby lowering the levels of pathogenic antibodies. It is evaluated as a treatment strategy applicable to autoimmune diseases in general. However, it appears to be struggling in the field of TED, beyond myasthenia gravis.HanAll Biopharma’s global partner, Immunovant, recently announced that its FcRn inhibitor batoclimab (IMVT-1401) failed to meet its primary endpoint in a global Phase 3 trial for TED. It failed to secure statistical significance in improving the Proptosis Responder Rate.Belgium-based Argenx also discontinued its clinical research on the FcRn inhibitor 'Vyvgart (efgartigimod)' for TED. Development was halted as the Independent Data Monitoring Committee (IDMC) judged the possibility of meeting the primary endpoints to be low.Accordingly, expert analysis suggests that a simple approach of reducing autoantibodies alone may be difficult to achieve sufficient clinical effects in TED.Chase by Viridian and Roche…the competition continuesNew drug development has not stagnated entirely. Among the followers, the most advanced is U.S.-based Viridian Therapeutics. Viridian is developing 'elegrobart,' a candidate based on the same IGF-1R mechanism as Tepezza.Elegrobart recently confirmed improvements in proptosis and diplopia in a global Phase 3 trial for patients with chronic TED, demonstrating the fastest rate of improvement among follow-up candidates.Roche’s 'Enspryng' However, Amgen has also moved to defend its market, recently reporting positive results from a Phase 3 trial of a subcutaneous (SC) formulation of Tepezza using an on-body injector (OBI). Consequently, competition in the TED market appears to be shifting toward competing for both efficacy and convenience, rather than simply SC conversion.Competition based on other mechanisms is also continuing. Roche’s 'Enspryng (satralizumab)' is a treatment that blocks the Interleukin-6 (IL-6) receptor, which is involved in inflammatory responses.Enspryng differs from the IGF-1R class, which directly targets ocular tissue expansion, by targeting the disease by suppressing orbital tissue inflammation and immune responses in TED.Roche recently announced positive results from the global Phase 3 SatraGO program for TED and disclosed plans to apply for authorization within the year.Enspryng failed to achieve statistical significance for the primary endpoint in the global Phase 3 SatraGO-1 study in patients with active TED. However, a pooled analysis with the follow-up Phase 3 study, SatraGO-2, reportedly confirmed a proptosis improvement effect.Until now, the treatment with the clearest clinical efficacy in TED has been the IGF-1R-targeted therapy. As FcRn inhibitors face successive clinical struggles, follow-up competition is trending toward a reorganization around IL-6R inhibitors and next-generation IGF-1R agents.

- Policy
- Government support for the next Leclaza discovery continues
- by Lee, Jeong-Hwan May 06, 2026 03:29pm
- Kang-seop Lim, Director of the Pharmaceutical Bio Industry Division, MOHW“A new drug candidate from the biotech venture Genosco was licensed out to Johnson & Johnson in the U.S. via the mid-sized pharmaceutical company Yuhan Corporation, leading to the creation of Leclaza. This is a prime example of a successful pharmaceutical and biotech startup from Korea. We plan to continue pushing forward with government policies this year to provide full support until the end, ensuring that more startup models emerge, with policies that support the commercialization of such early-stage discoveries.”The Ministry of Health and Welfare is unveiling a specialized startup fostering roadmap for the pharmaceutical and biotech sector for the first time in history, aiming to create domestic blockbuster drugs like a “second Leclaza” from the venture stage.The plan will provide full-cycle support to ensure promising drug candidates are not lost, linking technologies from universities, hospitals, and research institutes to startups, scaling them up, and supporting global expansion.On the 3rd, Kang-seop Lim, Director of the Pharmaceutical Bio Industry Division, stated that a startup support plan will be developed jointly with the Ministry of SMEs and Startups (MSS) and announced by June or July.With the newly established division within MOHW and Director Lim dedicating his full efforts as its first head, there is a growing momentum to develop multifaceted policies for the promotion of the pharmaceutical and biotechnology industry.This initiative follows President Jae-myung Lee’s ‘Startup Nation’ strategy announced in January.At that time, President Lee instructed all government ministries to build a startup ecosystem where anyone, including young people, regional entrepreneurs, and deep-tech startups, can pursue entrepreneurship.The MOHW is joining forces with the MSS on the plan, following their earlier announcement of collaborative measures for pharmaceutical and biotech industry policies.This amounts to establishing specific policy measures to promote and foster startups in the pharmaceutical and biotech industry, with the revitalization of startups in this sector serving as the policy goal and overarching framework.Director Lim pointed to research institutes within universities, medical institution research centers, including those in university hospitals, and government-funded research institutes as the specific targets of this policy.This administrative initiative aims to increase the number of cases where new drug candidates currently being researched by professors, doctors, and scholars are brought to the forefront through government-supported startups, commercialized and productized through processes such as licensing out, and then expanded beyond the domestic market into the global market.Director Lim cited Leclaza as a representative example. He explained that this is a policy in which the MOHW and MSS joined forces to ensure that a highly marketable domestic new drug, capable of succeeding in both domestic and overseas markets, was developed through the Genosco-Yuhan Corporation-Johnson & Johnson track.Director Lim stated, “Lecraza is a representative success case in which an early-stage drug candidate developed by Genosco was licensed by the mid-sized pharmaceutical company Yuhan, and later out-licensed to Johnson & Johnson for commercialization in overseas markets. This is a prime example of a successful pharmaceutical and biotech startup, and the government plans to support more cases like this.”He added, “In Korea, pharma-biotech startups typically emerge through several routes, including ventures founded by professors and researchers in university labs, physicians in hospitals, and researchers at government-funded institutes. A significant number of new drug candidates are likely to originate at the researcher or academic stage. The policy focus is on how the government can support these efforts when they transition into pharma-biotech startups.”Lim continued, “Given the limited size of the domestic market, global expansion is inevitable. We will support pharma-biotech startups not only in scaling up after their establishment, but also through to global expansion and commercialization.”He further stated, “This is the first time the Ministry of Health and Welfare has introduced a policy specifically targeting pharma-biotech venture startups. While we are still working through the details and identifying actionable support measures, we plan to finalize and announce the startup support plan by July. Ultimately, our goal is to establish a Ministry of Health and Welfare policy that identifies and supports startups that will serve as the seeds for large-scale innovative drug development.”

- Policy
- Remote delivery of medical supplies for rare disease patients allowed
- by Lee, Jeong-Hwan May 06, 2026 03:29pm
- Minister Eun-kyeong Jeong ㅆhe Ministry of Health and Welfare announced that starting on the 4th, it will implement a “non-face-to-face direct delivery service for medical products” targeting rare disease patients, patients with severe intractable diseases, and severely ill children.This is to ensure stable access to medical products for patients with rare diseases who are facing difficulties in securing necessary medical supplies due to the prolonged Middle East conflict.Target conditions include short bowel syndrome, Cornelia de Lange syndrome, Pompe disease, and biliary atresia. Medical products (including pharmaceuticals and medical devices) in high demand for each disease group, such as syringes and IV lines, are included in the remote delivery program.The Ministry will provide one-stop support through the telemedicine platform ‘Soldoc,’ which allows patients to consult with representatives via chat and make product purchases and delivery after patients and caregivers go through the verification process.In the future, the model will expand to include customized in-person and remote care, collaboration between large and local hospitals, and delivery of pharmaceuticals and medical supplies.On the 3rd, Minister Eun-kyeong Jeong held a roundtable meeting at Seoul National University Hospital Rare Disease Center with representatives from the Korean Organization for Rare Diseases, SNUH medical staff, and the Soldoc platform to discuss the initiative.During the meeting, participants shared challenges faced by rare disease patients due to the Middle East conflict, and decided to immediately launch a direct delivery service for medical supplies in collaboration with the telemedicine platform Soldoc.Patients with rare diseases are defined as those suffering from rare conditions affecting 20,000 people or fewer, as stipulated by the Rare Disease Management Act. Among these patients, those who must manage their conditions at home using medical supplies such as syringes and IV sets are facing difficulties due to rising prices and shortages of medical supplies caused by the war in the Middle East.In fact, Mr. A, a caregiver for a patient with short bowel syndrome, expressed his anxiety, saying, “I was worried because the IV sets I used to buy online were often out of stock due to the situation in the Middle East.”Ms. B, who cares for a child with Cornelia de Lange syndrome, also said, “Syringes and disposable vials are essential for the children’s nutritional support (enteral feeding) and medication administration, so I was worried I wouldn’t be able to obtain the supplies I usually buy from online shopping sites.”Ultimately, the difficulties in securing medical supplies purchased through online shopping sites due to the fallout from the Middle East conflict led to a partnership between the Ministry of Health and Welfare and the telemedicine platform SolDoc.Unlike general online shopping sites, Soldoc has a system in place to verify eligibility, being linked with medical institutions to confirm whether a patient has a rare disease.Through this system, when a patient with a rare disease or their caregiver submits a purchase request via the internet or app, eligibility verification is easily conducted through the National Health Insurance Service system.Once verified, they can purchase products and receive delivery, paying out-of-pocket for non-reimbursed items.For items covered under medical expense reimbursement that require prescriptions, patients can consult doctors via telemedicine before purchase. Claims are handled by the provider, and patients only pay their coinsured share.Items available include syringes, infusion sets, suction tips, suction catheters, sterile saline, and disinfectant swabs.The Ministry plans to expand the program to include patients with severe intractable diseases and children receiving medical expense support, if necessary.Additionally, the Ministry is considering pharmaceutical delivery for urgent cases.Telemedicine will be formally implemented in December, following revisions to the Medical Service Act. The revised law allows telemedicine for rare disease patients.In particular, patients with rare diseases can receive telemedicine services even at hospital-level or higher medical institutions, and the delivery of drugs and supplies is also permitted. The Ministry plans to strengthen services focused on those requiring essential medical services through telemedicine before the law takes effect.Minister Eun-kyeong Jeong promised, “The state and society will take responsibility to ensure that patients are not marginalized or left in anxiety simply because their diseases are rare. We will provide financial support for the cost of medical supplies if needed after reviewing the burden of medical supply costs.”Meanwhile, on the same day, Minister Jeong, Soon-heon Kwak, Director General of Health and Medical Policy, Jin-hyang Jeong, Secretary General of the Korean Organization for Rare Diseases, seven patients, and six medical staff members, including Joong-shin Park, Vice President for Medical Services at Seoul National University Hospital, held a staff meeting at Seoul National University Hospital in Jongno-gu, Seoul, after the roundtable meeting.
- Company
- A year after Leclaza securing European approval…Yuhan
- by Chon, Seung-Hyun May 06, 2026 03:28pm
- Yuhan expects to receive a $30 million technology fee payment for the European market entry of its new anticancer drug, Leclaza, soon.During the recent Q1 business performance presentation, Yuhan stated, “We expect to receive the $30 million milestone associated with the European launch of the lazertinib combination therapy soon,” and added, “The business plan established early this year is progressing smoothly.”Lazertinib, the active pharmaceutical ingredient of Leclaza, is a new anticancer drug developed by Yuhan. Leclaza is a non-small cell lung cancer (NSCLC) treatment that was approved in January 2021 as the 31st domestically developed new drug in Korea.In December 2024, the European Commission (EC) approved the combination therapy of Leclaza + Rybrevant as a first-line treatment for adult patients with locally advanced or metastatic NSCLC harboring epidermal growth factor receptor (EGFR) exon 19 deletions or exon 21 L858R substitution mutations.While the $30 million (approx. KRW 44 billion) milestone was met with Leclaza's entry into the European market, the payment has not yet been received, despite 17 months having passed. According to the company, the milestone will be transferred once sales of Leclaza begin in major European countries. Since the beginning of this year, Leclaza has been listed for health insurance in countries such as the UK, Switzerland, Italy, and Germany, and sales have begun. Securities analysts anticipate the European technology fee will be received within the first half of the year.Yuhan recorded KRW 5 billion in technology fee revenue in the first quarter. This is a 23.7% increase from the KRW 4 billion recorded in Q1 of last year, but significantly lower than the KRW 70.3 billion recorded in the previous quarter. Technology fee revenue inherently fluctuates, as it is generated from new drug licensing agreements or the advancement of development stages for licensed-out drugs.Last year, Yuhan received KRW 104.1 billion in technology fee revenue, with milestones from approvals in Japan and China accounting for a large portion.In May of last year, as the Japanese Ministry of Health, Labor and Welfare approved the Leclaza + amivantamab combination therapy, the requirement for an additional $15 million milestone was met, resulting in KRW 25 billion in technology fee revenue in Q2 of that year.In Q4 of last year, KRW 70.3 billion in technology fees were received. In August last year, China's National Medical Products Administration (NMPA) approved Leclaza as a combination therapy with Rybrevant, and Yuhan received a $45 million (KRW 69 billion) milestone from its partner, Janssen Biotech, for achieving the step-by-step milestone for Leclaza.Yuhan's Q1 technology fee revenue includes royalties from Janssen's sales of Leclaza. In 2024, the Leclaza and Rybrevant combination therapy received U.S. FDA approval, and sales in the U.S. began. According to Johnson & Johnson's performance data, Q1 sales of the Leclaza-Rybrevant combination therapy reached $257 million (approx. KRW 300 billion), an 82.7% increase year-on-year.Yuhan stated, “Prescription of the Leclaza combination therapy is expanding in major markets such as the U.S. and Europe, and we have secured a foundation for full-scale revenue growth through its listing as a 'Preferred Treatment' in the NCCN guidelines and the approval of the Rybrevant SC formulation.”In the 2026 National Comprehensive Cancer Network (NCCN) guidelines for NSCLC, the Leclaza + Rybrevant combination therapy was included as a 'Preferred Treatment' for first-line treatment. Leclaza is the first domestically developed new drug to be incorporated into the NCCN Category 1 first-line treatment.Yuhan received a steady inflow of technology fee revenue since 2018, when it began licensing new drug technologies.In July 2018, Yuhan licensed the technology for the degenerative disc disease treatment YH14618 to Spine BioPharma in the U.S. It received an upfront payment of $650,000 and was guaranteed $217.5 million in step-by-step milestones based on development, approval, and sales.In November 2018, Yuhan licensed out the anticancer drug Leclaza to Janssen Biotech. The total contract size, including a non-refundable $50 million upfront payment, is up to $1.205 billion.In January 2019, Yuhan signed a license and co-development agreement with Gilead Sciences for a new drug candidate targeting two drug targets for the treatment of metabolic dysfunction-associated steatohepatitis (MASH). The terms included a $15 million upfront payment and $777 million in milestones based on development, approval, and sales.In July 2019, Yuhan signed a technology transfer agreement for YH25724 with Boehringer Ingelheim. YH25724 is a dual-agonist targeting the GLP-1 protein and FGF21 factor simultaneously, with the technology transfer agreement signed during the preclinical stage. For this contract, Yuhan received a non-refundable upfront payment of $40 million. An additional $10 million milestone was generated for YH25724 in November 2021 upon entering Phase 1 clinical trials.In August 2020, Yuhan signed a technology transfer agreement with Processa Pharmaceuticals in the U.S. for YH12852, a treatment candidate for functional gastrointestinal disorders, in which Yuhan received a non-refundable $2 million upfront payment in the form of stock. Yuhan recognized the upfront payments and milestones received from the other four companies excluding Processa, which paid in stock, in installments. Yuhan received total technology fee revenue in 2019 until the first quarter of this year is amounted to be KRW 465 billion. Of the Leclaza technology fee revenue secured by Yuhan, 40% is paid to the original developer, Oscotec. In 2016, Yuhan acquired the development rights for Leclaza at the preclinical stage from Oscotec and its subsidiary Genosco. The total contract size for that acquisition was KRW 1.5 billion.

- Policy
- Incrementally modified drugs unaffected by price reform
- by Jung, Heung-Jun May 06, 2026 03:28pm
- Under the government’s drug pricing system reform, the insurance pricing premium rates for incrementally modified drugs and their combination products are expected to remain unchanged. Only some conditions related to the duration of the premium are likely to be adjusted.As the pricing calculation rate for generics is set to drop to 45%, domestic companies are expected to show greater interest in developing incrementally modified drugs.According to industry sources on the 6th, during working-level discussions between the government and the pharmaceutical industry, a consensus was reached not to lower the premium for incrementally modified new drugs.Although maintaining the current premium was discussed at the Health Insurance Policy Deliberation Committee (HIPDC) in November last year, the point was excluded from the reform plan approved by the HIPDC in March this year.This led to concerns within the industry that the premium might also be reduced along with the lower pricing calculation rate. There were concerns that lowering the premium could eliminate incentives for R&D investment.Under the current pricing system, incrementally modified drugs receive a premium on a base pricing rate of 53.55%, resulting in a final price of 70%. For new dosage or administration forms, a 58.9% premium is applied, resulting in a price of 77%.For incrementally modified combination drugs, pricing is calculated as the sum of pre-patent-expiry prices of each component. Innovative pharmaceutical companies receive 68% of that sum, while general pharmaceutical companies receive 59.5%.The government is not expected to significantly adjust these premium rates. Instead, it is reported that the preferential premium rate for combination drugs by general pharmaceutical companies—currently a 59.5% sum—will be slightly adjusted to a 60% sum.As the generic pricing rate is reduced from 53.55% to 45% while the incrementally modified drug premium remains unchanged, the price gap between generics and modified drugs is expected to widen further.The conditions for the premium duration are expected to be simplified. Previously, a one-year premium could be extended in two-year increments through conditional approvals and reviews.Going forward, a basic one-year premium will be granted, with an additional three-year extension if domestic manufacturing criteria are met. If no follow-on generics are listed thereafter, the premium may continue.Modified drugs and their combination products that are domestically produced and face no market competition will be able to maintain their premium drug prices for a long time.
- Opinion
- [Reporter's View] Contradiction of "K-passing" and a new drug powerhouse
- by Lee, Jeong-Hwan May 06, 2026 03:28pm
- The President Lee Jae Myung administration is promoting the growth of the pharmaceutical and biotech industry with goals of 'Rising as a leading country in global pharmaceuticals,' 'Strengthening treatment accessibility for patients with severe and rare/intractable diseases,' and 'Expanding fair value compensation for innovative new drugs.'The government's stance on the end goal of the drug pricing system reform plan, which recently passed the Health Insurance Policy Review Committee, is to transform the inherent nature of Korea's pharmaceutical and biotech industry, aiming to 'develop new drugs·stably supply essential medicines·expand patient accessibility to reimbursement.'Despite the government's policy vision, South Korea is facing "New Drug Korea-Passing." The South Korean government is not immune to the phenomenon in which pharmaceutical companies that have developed innovative new drugs delay or abandon their launches in the Korean market.While "New Drug Korea-Passing" has long been practiced primarily by global pharmaceutical companies, the rapid improvement in domestic firms' drug development capabilities suggests a future in which Korean deciding to bypass Korea. One of these examples is Cenobamate (brand name Xcopri), a new epilepsy drug developed by SK Biopharmaceuticals and Dong-A ST.It is a contradiction that South Korea, while seeking to become a global pharmaceutical powerhouse, must now worry about the availability of patient treatment, given the "Korea-Passing" phenomenon.The bigger issue is that it is difficult to find any serious deliberation at the government level to establish a solution.The cause of "New Drug Korea-Passing" is "low National Health Insurance (NHI) reimbursement prices for new drugs." Criticism follows that Korea's maximum reimbursement prices for new drugs are only half the OECD average and about 1/30 of those in the United States.When pharmaceutical companies accept Korea's low drug prices, other countries may use them as a reference, leading companies to abandon the relatively small Korean market. Ultimately, the victims of these decisions are the patients who must bear the full burden.The reason the South Korean government tries to set new drug prices as low as possible is not entirely incomprehensible. Since they attempt to set prices using the NHI fund, composed of citizen contributions, as the sole source of financing, it is inevitably difficult to determine a price that fully reflects the value of an innovative drug.Ultimately, the conclusion is reached that to realize a pharmaceutical and biotech powerhouse and solve the "New Drug Korea-Passing" problem, substantial financial resources are required to set prices that reflect the proper value of new drugs.This means that government efforts to secure separate financial resources outside of the NHI fund to determine new drug prices are needed immediately. The solution to achieving both the conflicting tasks of securing the sustainability and soundness of NHI finances while strengthening patient access to medicines also involves breaking away from the single-source NHI funding structure.The consequence of failing to manage the national task and securing separate funds has consistently manifested as a reduction in pharmaceutical spending through generic drug price cuts, repeated in the same pattern every time. This is why criticism arises that, while intense strategic posturing continues between the Ministry of Health and Welfare (MOHW), global pharmaceutical companies, domestic pharmaceutical companies, and patient groups over how to distribute limited resources, only innocent generic companies are hit.Unless the structure that relies entirely on the NHI fund to expand innovative drug reimbursement, amid an era of super-aging and the increasing launch of ultra-expensive new drugs, is reformed, there is no place for the MOHW, the domestic pharmaceutical industry, or patients.Various methods for creating separate funds beyond the NHI can be discussed. These include establishing funds dedicated to ultra-expensive medicines, similar to the UK's Cancer Drugs Fund, or implementing policies that allow a portion of tobacco taxes or lottery proceeds to be used for innovative drug reimbursement.Legislative bills for such policies have been proposed in the National Assembly for over a decade. The key is the government's will. Responsibility should not be placed solely on the MOHW. It requires a policy decision from the Ministry of Economy and Finance, the Ministry of Planning and Budget. Furthermore, the Prime Minister and the President. Are they not the drivers who set the policy goals of leaping into a pharmaceutical and bio-tech powerhouse and strengthening patient access to new drugs?In the National Assembly, policy seminars calling for the rapid reimbursement of innovative new drugs and the expansion of reimbursed indications are held daily, and the heavy responsibility for solving the problem is habitually returned to the Bureau of Health Insurance Policy of the MOHW. Can we continue to demand a solution for the expansion of new drug reimbursement and the "Korea-Passing" problem from the MOHW alone?It is time for the fiscal authorities, besides the MOHW, to take the lead with proactive measures to solve the task of securing separate funds through social consensus, and to immediately resolve the contradiction where being a 'new drug powerhouse' and "Korea-Passing" coexist. The President's political slogan, "I'll do it," should not be an exception when it comes to strengthening access to innovative drug reimbursement and expanding financial resources.
- Policy
- What's the reason behind domestically developed CAR-T 'Rimqarto' obtaining Phase 3 waiver?
- by Lee, Tak-Sun May 04, 2026 10:33am
- CAR-T therapy Rimqarto (source: Curocell)Rimqarto (anbalcabtagene autoleucel, Curocell), the first domestically developed CAR-T therapy to be approved in South Korea, has been granted a waiver for Phase 3 clinical trials.This decision is interpreted as the result of a comprehensive consideration of the unique characteristics of the drug as a third-line treatment for lymphoma, as well as the ethical dilemmas associated with comparative clinical trials against existing therapies.According to the results of the Ministry of Food and Drug Safety (MFDS)'s Central Pharmaceutical Affairs Advisory Committee (CPAC) meeting held on April 2, it was concluded that it is appropriate to waive the Phase 3 clinical trial for the new CAR-T (Chimeric Antigen Receptor T-cell) therapy 'Rimqarto' and replace it with post-marketing surveillance.CPAC members highlighted that while Rimqarto has the same basic mechanism as existing CAR-T agents, it introduces a novel mechanism that simultaneously inhibits PD-1 and TIGIT to prevent T-cell exhaustion.According to a recently disclosed meeting report, one member highly evaluated the product's efficacy, stating, "The response rates were better than the clinical results of previously approved therapies, particularly with a high proportion of patients achieving complete remission (CR) and encouraging long-term survival results."Regarding safety, no specific issues were found besides the adverse events typically reported in similar agents (such as CRS and ICANS), and deaths during the trial were judged to have a low correlation with the drug.On the highly debated issue of giving a 'conditional pass for Phase 3 clinical trials,' the committee reached a consensus that it is "practically impossible." First, they viewed it as lacking ethical validity. Given that already-proven CAR-T products are approved and in use, administering a less effective control drug to patients was deemed unethical.The difficulty of patient recruitment was also considered. The patient population in the third-line lymphoma treatment phase has a low survival rate and a small number of candidates, making it extremely difficult to conduct large-scale confirmatory trials that include a control group. Consequently, the CPAC concluded that it is more rational to continuously verify safety and efficacy using Real-World Data (RWD) collected in clinical settings or Post-Marketing Surveillance (PMS), rather than mandating a Phase 3 trial.Based on this CPAC advisory, the MFDS finalized the approval conditions for Rimqarto. The committee agreed that the "product approval is appropriate, given that it is a third-line lymphoma treatment," and decided to disclose the meeting report anonymously.This decision served as a stepping stone toward the rapid supply of an independently developed domestic CAR-T therapy to the field. It is expected to provide new treatment opportunities for patients with severe hematologic cancers who do not respond to existing treatments.Meanwhile, 'Rimqarto Inj' is an orphan drug for the treatment of adult patients with diffuse large B-cell lymphoma (DLBCL) and primary mediastinal B-cell lymphoma (PMBCL) that is relapsed or refractory after two or more systemic therapies.Rimqarto works by inserting genetic information into the patient's immune cells (T-cells) to enable them to recognize CD19, a surface antigen on B-cells, and then re-injecting these cells into the patient's body to identify and destroy cancer cells expressing CD19. It is designed to inhibit the expression of immune checkpoint receptors PD-1 and TIGIT, thereby blocking cancer cells' immune evasion and inducing enhanced, sustained T-cell responses to increase anti-tumor effects.