- LOGIN
- MemberShip
- 2026-05-06 00:22:40

- Company
- Celltrion’s Steqeyma approved in CAN…targets Stelara mkt
- by Hwang, Byung-woo Aug 01, 2024 05:48am
- On the 31st, Celltrion announced that it had received New Drug Application (NDS) approval for its Stelara biosimilar Steqeyma from Health Canada. With the approval, Celltrion may now sell Steqeyma in Canada for plaque psoriasis, psoriatic arthritis, and Crohn's disease. The Canadian approval supports the company’s plan to settle into the global ustekinumab market starting with North America, the largest pharmaceutical market. According to IQVIA, the global ustekinumab market was valued at around USD 20.4 billion (KRW 26.52 trillion) last year, with the Canadian market recording USD 663 million (KRW 861.9 billion). When including the U.S. market, the total North American market is about $16.375 billion (KRW 21.285 trillion), accounting for more than 80 percent of the global market. Celltrion previously received approval for Steqeyma in Korea in June and a positive opinion for marketing authorization from the European Medicines Agency's (EMA) Committee for Medicinal Products for Human Use (CHMP). When approved, the European approval will further accelerate Celltrion's expansion into the global ustekinumab market. The approval will also expand Celltrion's autoimmune disease treatment portfolio in North America, adding an interleukin (IL) inhibitor to its existing tumor necrosis factor (TNF)-α inhibitor product line, expanding the target patient population. "Canada has pro-biosimilar policies in place that actively encourage the prescription of biosimilars, which will help Celltrion gain presence in the country," said a Celltrion official. "North America is the world's largest market for ustekinumab, and we expect the approval of Steqeyma in Canada to be the beginning of our expansion into the North American market." Meanwhile, Celltrion plans to secure a portfolio of 11 biosimilars by 2025, with the goal of strengthening its presence in the global market and increasing revenue.
- Company
- Will the cardiomyopathy drug Vyndamax be reimb this time?
- by Eo, Yun-Ho Aug 01, 2024 05:48am
- The reimbursement listing process for ‘Vyndamax,’ Pfizer’s treatment for transthyretin amyloid cardiomyopathy (ATTR-CM), is gaining attention in Korea. According to industry sources, discussions for the reimbursement of Pfizer Korea’s Vyndamax (tafamidis 61mg) for Transthyretin amyloid cardiomyopathy (ATTR-CM) have again begun. Once again, the key players will be the Pharmacoconomic Evaluation Subcommittee and the Reimbursement Standard Subcommittee. Vyndamax failed its first reimbursement attempt in early 2021. The company then made its second attempt in the first half of the same year after conducting a pharmacoeconomic evaluation and preparing a risk-sharing agreement (RSA) but saw the same results. Then, in April 2022, the drug failed to pass the Health Insurance Reimbursement and Assessment Service’s reimbursement subcommittee’s threshold again, then passed the threshold in July of the same year, but was again rejected by the Drug Reimbursement Evaluation Committee after 9 months. At the time, it was believed that the authorities failed to close the gap with the pharmaceutical company on the terms of the RSA. Therefore, it remains to be seen whether Vyndamax will be able to pass the DREC review and move on to the drug pricing negotiation stage this time. Vyndamax is virtually the only treatment option available for ATTR-CM. ATTR-CM is a fatal condition with a poor treatment outcome due to a lack of specific treatment and is often mistaken for simple heart failure If not treated properly, patients with ATTR-CM have a survival period of only 2 to 3.5 years. In this area with a dire need, Vyndamax demonstrated a reduction of cardiovascular events in ATTR-CM patients and improvement in their functional athletic ability in the six-minute walk through the Phase III ATTR-ACT study. Based on these results, healthcare professionals in Korea have been insisting on the need to prescribe Vyndamax. “The approval of Vyndamax last year has greatly advanced Korea’s ATTR-CM treatment landscape, bringing substantial survival benefits for patients with ATTR-CM,” said Jung-Woo Son, Professor of Cardiology at Wonju Severance Christian Hospital. “However, the drug is still not reimbursed in Korea, and patients are left unable to start treatment even after diagnosis.”
- Opinion
- [Reporter’s View] More legislation needed for CSO reporting
- by Lee, Jeong-Hwan Aug 01, 2024 05:48am
- The enforcement of the Pharmaceutical Affairs Act, which requires Contract Sales Organizations (CSOs) to report to local governments and educate their employees on the prohibition of rebates, is now 2 months away. The pharmaceutical and CSO industries are hoping the implementation of the reporting system will serve as a milestone to increase the transparency of illegal drug rebate practices and put behind their past where CSOs were used as black money payment channels to promote the physicians’ prescription of certain drugs. In law with the law's implementation, the Ministry of Health and Welfare (MOHW) announced the proposed amendments to the Enforcement Rules of the Pharmaceutical Affairs Act on March 18 and began collecting industry opinions. Once the reporting system is implemented, CSOs are expected to ensure justice in sales practices within the pharmaceutical and biotechnology industry and be incorporated into the system, thus being manageable by the government. Of course, the Ministry of Health and Welfare, the pharmaceutical industry, and the CSO industry will need to work together to improve the problems or shortcomings that may arise upon the implementation of the system, but the CSOs, whose existence has been virtually unrecognized until now, will be able to take shape and advance the pharmaceutical industry’s sales practices. However, there is still legislation that needs to be enacted in line with the CSO reporting system -the Medical Service Act. The act needs to prohibit doctors from accepting rebates, including financial kickbacks and entertainment provided by CSOs. Such amendment to the Medical Service Act passed the Health and Welfare Committee review and was pending at the Legislation and Judiciary Committee’s 2nd Bill Review Subcommittee stage at the 21st National Assembly, but was eventually abandoned due to the closure of the 21st NA. The CSO reporting system and training obligations will strengthen the government and local governments' control over CSOs' illegal rebate practices, but there are concerns that its effect may be halved unless it is followed by an amendment to the Medical Service Act, which will prohibit doctors from accepting rebates. Article 23(5) of the Medical Service Act, which prohibits doctors from gaining improper financial benefits, needs to be amended. The "drug suppliers" need to be changed to "drug suppliers and contract sales organizations (CSOs)" to provide a clear legal basis to punish doctors for accepting rebates from CSOs. Some have even suggested that amending the Medical Service Act would be more effective in eradicating rebates than the CSO reporting system. Therefore, the Ministry of Health and Welfare and the 22nd National Assembly should work together to promptly amend the Medical Service Act. The authorities need to minimize the period of the CSO reporting system being operated as a half-baked system because the legal basis for prohibiting doctors from receiving rebates for CSO payments has not been established. The MOHW should reiterate the need to quickly introduce, examine, and pass the proposed amendments to the Medical Service Act, and the ruling and opposition parties in the Health and Welfare Committee should also join forces to expedite the passage of such legislation to achieve the goal of eliminating rebates. With the CSO reporting system set to go into effect on October 19th, the National Assembly needs to seriously consider passing amendments to the Medical Service Act to enhance the effectiveness of the system and fill in the gaps in the legislation in time.
- Company
- Kim Sang-pyo, appointed as Moderna Korea's new GM
- by Eo, Yun-Ho Aug 01, 2024 05:47am
- Kim Sang-pyo, appointed as the new General Manager of Moderna Korea. Moderna Korea appointed Kim Sang-pyo, the former head of AstraZeneca Korea, as the new General Manager. Moderna announced the appointment of Kim Sang-pyo as the new General Manager, effective on August 1st. Kim will head Moderna's subsidiary in South Korea, succeeding Sohn Ji-young, who led the company's establishment in the Korean market and overall business operations since 2021. Kim stated, "South Korea is an important market for Moderna's global management strategy. I am deeply responsible for further strengthening Moderna's leadership in mRNA technology. Furthermore, I will realize Moderna's mission, which is to bring out the potential of mRNA medicines, and strive to contribute to the nation's healthcare." Kim is an expert who has accumulated experience and achievements working in the pharmaceutical industry for over 20 years. He has been the head of AstraZeneca from 2018 until recently, achieving the goals of promoting management efficiency and growth, maximizing new product launch opportunities, and developing global business projects, such as COVID-19 vaccine distribution. Before AstraZeneca, he held leadership positions in various domestic and global business sectors, including oncology general manager and business unit director at MSD. Kim led business growth and innovation, earning recognition for his role and leadership. Patrick Bergstedt, Senior Vice President of Asia and Emerging Markets at Moderna, said, "We welcome Kim's appointment as the new GM. With his extensive experience and deep insights into the healthcare industry, we look forward to having Kim lead Moderna Korea." Since its official launch in June 2021, Moderna Korea has contributed to overcoming the pandemic by introducing the COVID-19 vaccine to South Korea. The company has also established partnerships with the Korean government and research institutes and will use the mRNA platform technology to help solve various healthcare issues. This fall, Moderna Korea will supply updated COVID-19 vaccines in alliance with the government's plan to protect the nation from COVID-19 mutations.

- Company
- Combination cancer immunotherapies show high synergy
- by Son, Hyung-Min Aug 01, 2024 05:47am
- Data demonstrating the effectiveness of the combination of cancer immunotherapies are being disclosed. Recently, Opdivo+Yerboy therapy has been shown to improve the survival duration when used as a first-line treatment of hepatocellular carcinoma (HCC). GI Innovation confirmed partial response (PR) using cancer immunotherapies as combination therapy in clinical trials. TiumBio also observed cancer cell death in various solid tumors, including pancreatic cancer, anal cancer, and lung cancer. AptaBio is assessing the potential of cancer immunotherapy Keytruda as a combination therapy. According to industry sources on July 31st, Bristol Myers Squibb (BMS) has recently applied to the European Medicines Agency (EMA) for approval of Opdivo+Yerboy combination therapy for the first-line treatment of HCC. Optivo is an immunotherapy for cancer treatment jointly developed by BMS and Ono Pharmaceutical, and it targets PD-1/PD-L1. Optivo is currently being studied for potential use in various solid tumors in combination with CTLA-4 targeting immunotherapy Yerboy. Cancer immunotherapies, Optivo and Yervoy.Opdivo+Yerboy combination therapy was previosly approved as a first-line treatment of renal cell carcinoma, and it is being studied for potential use in various fields, including colorectal cancer, esophageal cancer, and HCC. The company has applied for an expanded indication to the EMA for the use of combination therapy as a first-line treatment of renal cell carcinoma. Although various treatments are already available in the market, such as Avastin+Tecentriq and Imfinzi+Imjudo, different combination therapies, including Opdivo+Yerboy and rivoceranib plus camrelizumab, are challenging the market with new data. The basis of approval was the Phase 3 CheckMate-9DW trial. In the clinical trial, 668 patients with liver cancer who have not received prior treatment were randomized to receive either Opdivo+Yerboy combination therapy or Lenvima+Nexavar therapy. The clinical result showed that patients treated with Opdivo+Yerboy combination therapy had a median overall survival (OS) of 23.7 months, demonstrating improvement compared to the control group's 20.6 months. The objective response rate (ORR) for Opdivo+Yerboy combination therapy was 36%, whereas it was 13% for the control group. The median duration of response (DOR) for Opdivo+Yerboy combination therapy was 30.4 months, longer than the 12.9 months of the control group. The clinical results served as a foundation for Opdivo+Yerboy combination therapy to gain a competitive advantage in the market as a first-line treatment of liver cancer. Korean pharma & biotech companies' combination cancer immunotherapies have shown potential in clinical trials Korean pharmaceutical and biotech companies are evaluating the potential commercialization of combination cancer immunotherapies. TiumBio recently disclosed its data on TU2218. TU2218 blocks pathways of transforming growth factor beta (TGF-ß) and vascular endothelial growth factor (VEGF), which are known to hinder cancer immunotherapy activation. TU2218's mechanism maximizes the efficacy of cancer immunotherapy. TiumBio is conducting a Phase1b trial in three clinical institutes in the United States to evaluate the efficacy and safety of TU2218 in combination with Keytruda in patients with advanced solid tumors. During the Phase 1b trial, TiumBio confirmed partial response (PR) from two patients and stable disease (SD) from three patients out of five patients who are evaluable for efficacy. Also, TU2218 showed an ORR of 40% and a disease control rate (DCR) of 100%. Additionally, lung cancer patients had PR in addition to previously shown PR results in patients with pancreatic cancer and anal cancer. In the TU2218 clinical 1b trial, three patients were found to have PR to date. GI Innovation recently disclosed its data on a Phase 1/2 trial of GI-101A plus Keytruda combination therapy. GI-101A's mechanism involves CD80 and interleukin (IL)-2. IL-2 is involved in immune cell proliferation and activation, and CD80 plays a role in blocking CTLA4, a receptor inhibiting immune cells that attack cancer cells. GI-101A is a new drug candidate developed by increasing the sialic acid content of GI-101 during manufacturing, thereby prolonging the safety and half-life. GI Innovation is conducting the Phase 1/2 trial of GI-101A in combination with Keytruda in South Korea and the United States. When GI-101A+Keytruda combination therapy was administered in patients with pancreatic cancer, who have liver metastasis and had failed prior chemetherapy, the pathology was reduced by 73%. GI-101A+Keytruda combination therapy has also been effective in reducing pathology in patients with renal cell cancer. When GI-101A+Keytruda combination therapy was administered to patients who failed 10 oncology treatments, including cancer immunotherapy, the pathology was reduced by 39%. AptaBio is assessing the potential of cancer immunotherapy candidate AB-19 as monotherapy and cancer immunotherapies AB-19+PD-1 as combination therapy. AB-19's mechanism is designed to block cancer-associated fibroblasts (CAF) within the tumor microenvironment of cancer cells. In a preclinical study, AB-19 monotherapy and cancer immunotherapy as combination therapy were shown to have superior effects on reducing tumor sizes than conventional PD-1 inhibitors. In April, AptaBio completed registering AB-19's patent in the United States, following in Russia and Australia. The company is awaiting for patent registration in Japan and China.

- Company
- Commercialization of global RSV vaccines in progress in KOR
- by Son, Hyung-Min Jul 31, 2024 05:52am
- A global respiratory syncytial virus (RSV) vaccine has entered late-stage clinical trials for the first time in Korea, testing its potential for commercialization. Pfizer recently received approval to initiate a Phase III clinical trial in Korea to evaluate the immunogenicity of its RSV vaccine. Moderna is applying for Japanese approval of its RSV vaccine, which was approved in the U.S. in May, and making its way into Asian countries. Among domestic biotech companies, EuBiologics has entered Phase I clinical trials in Korea. RSV is a virus that causes pneumonia and bronchitis. The virus can affect a person of any age, but the infection rate is particularly high in infants and young children. It is the leading cause of hospitalization in infants and young children, and 90% of infants worldwide are infected with RSV before the age of 2. When infected, RSV causes symptoms similar to the common cold, but some infants and children may experience a more severe course, leading to lower respiratory tract illnesses such as pneumonia and bronchiolitis. According to industry sources on the 30th, the Ministry of Food and Drug Safety approved the Phase III IND for the RSV vaccine 'PF-06928316' submitted by Pfizer on the 26th. The trial will be conducted at 16 institutions, including Samsung Medical Center, Severance Hospital, and Seoul St. Mary's Hospital, from October this year to December next year. In the trial, Pfizer will evaluate the safety, tolerability, and immunogenicity of the RSV vaccine in 360 older adults. The primary endpoints will be local reactions (redness, swelling, and pain at the injection site) and systemic events (fever, fatigue, headache, vomiting, nausea, diarrhea, muscle pain, and joint pain), based on which the company will evaluate the adverse events and tolerability of PF-06928316. Participants will be randomized in a 2:1 ratio to PF-06928316 or placebo. PfizerPfizer’s RSV vaccine was approved in the U.S. last year and launched under the brand name Abrysvo. Abrysvo is indicated for adults 60 years of age and older, infants 6 months of age and younger, and pregnant women at less than 37 weeks gestation. Recently, Pfizer confirmed the immunogenicity of Abrysvo in adults aged 18 to 59 years, raising the possibility of its indication expansion. Abrysvo demonstrated non-inferior neutralization responses in 681 adults aged 18 to 59 years with certain chronic diseases and 200 immunocompromised adults compared to that identified in the original clinical trial that evaluated the vaccine in older adults. Participants also achieved at least a four-fold increase in serum neutralizing titers following receipt of Abrysvo compared to pre-vaccination. These results support the potential for Abrysvo’s expanded indication. It also opened the door for Pfizer to enter clinical trials in Korea to evaluate the immunogenicity of the RSV vaccine in all ages. Moderna receives approval in the U.S. and seeks to enter the Asian market…EuBiologics starts clinical trial in Korea The RSV vaccine market is currently in a three-way battle between Abrysvo, GSK’s ‘Arexvy,’ and Moderna’s mRNA-1345, with Moderna conducting a clinical trial to develop a follow-on drug. Among the companies, Moderna has applied for mRNA-1345’s approval in Japan and is seeking to enter the Asian market. Moderna submitted an application for mRNA-1345 to Japan's Ministry of Health, Labour and Welfare in May. mRNA-1345 was approved in the U.S. in May and was developed to prevent RSV in adults aged 60 and older. The application is based on the interim analysis results of the Phase III ConquerRSV that demonstrated immunogenicity and tolerability of mRNA-1345. The trial was conducted in 22 countries, including Japan. The ConquerRSV trial was a randomized, double-blind, placebo-controlled study that enrolled approximately 37,000 adults aged 60 years and older in 22 countries, including the United States. The interim analysis was based on two definitions of RSV lower respiratory tract disease (RSV-LRTD), defined as two or more symptoms or three or more disease symptoms. Results showed that in RSV-LRTD defined as two or more symptoms, mRNA-1345 met all primary endpoints, including demonstrating 83.7% vaccine efficacy. Adverse events with mRNA-1345 were mild to moderate, with the most commonly reported adverse events in the mRNA-1345 arm being injection site pain, fatigue, headache, myalgia, and arthralgia. In addition to the U.S. and Japan, Moderna plans to file for approval of mRNA-1345 in the European Union (EU) and other countries. In Korea, EuBiologics has entered clinical trials. The euRSV vaccine being developed by EuBiologics has recently been administered to 3 RSV patients at Korea University Guro Hospital. Patients were randomized to placebo, low-dose EuRSV, and high-dose EuRSV to evaluate the vaccine’s tolerability and safety. EuRSV is a recombinant protein subunit vaccine that combines the virus’s F-protein antigen with an immune booster. In 2017, Eubiologics in-licensed the immune booster EuIMT from the Korea Institute of Science and Technology. EuBiologics aims to enter global Phase 2/3 trials through its U.S. subsidiary, Eupop Life Sciences, which holds the marketing rights for EuRSV in the North American and European markets. In addition, SK Bioscience is also developing an mRNA-based RSV vaccine candidate like that being developed by Moderna. SK Bioscience began RSV vaccine development in 2018 with the aim to commercialize it by 2029 but is still in the basic research stage.

- Policy
- Approval of abortion drugs at a halt in KOR
- by Lee, Jeong-Hwan Jul 31, 2024 05:52am
- The Ministry of Food and Drug Safety has gained attention for stating that the Criminal Code and the Mother and Child Health Act must be amended before the approval of abortion drugs in Korea. The process of gaining marketing authorization for abortion drugs that the pharmaceutical companies applied for has been at a halt for 4 years, as follow-up legislation has not been enacted since the constitutionality of abortion was ruled unconstitutional. On the 29th, the Ministry of Food and Drug Safety responded so to a point raised by Representative Sun-min Kim of the National Assembly's Health and Welfare Committee regarding the domestic approval of abortion drugs. Kim raised the need for expedited approval of abortion drugs, questioning why the abortion drug has not yet been approved despite repeated applications submitted in 2021 and 2023. Hyundai Pharm applied for the marketing authorization of Mifegymiso in 2021 after signing an exclusive marketing and distribution agreement for its supply in Korea with the UK-based Linepharma International. However, the company voluntarily withdrew its application upon the MFDS’s data supplement request. The company then reapplied for the license last year (2023) and received another data supplementation request. Mifegymiso is a combination pack that contains 1 200mg mifepristone tablet and 4 misoprostol 200ug tablets. The drug inhibits the action of progesterone, the progestational hormone that sustains pregnancy, while misoprostol works to contract the uterus. It is only indicated for use in the first trimester of pregnancy, up to 9 weeks, and is typically prescribed as one tablet of mifepristone followed by 4 tablets of misoprostol 36-49 hours later. The MFDS’s position is that the Criminal Code and the Mother and Child Health Act must be amended before Mifegymiso, which the company has reapplied for approval, can be approved in Korea. The logic is that the abortion period should be established first so that a risk management plan can be submitted for the approval of abortion drugs like Mifegymiso. "Currently, there are permit data that can only be submitted after the amendment of the Criminal Code and the Mother and Child Health Act, which is why the review process has been temporarily suspended upon mutual recognition of the applicant and the MFDS,” said an MFDS official. "If the relevant laws are amended and the applicant submits data accordingly, we plan to resume the review without delay and decide whether to approve or reject the application.” Meanwhile, when the amendments will be made to the Criminal Code and Mother and Child Health Act remains unclear. In April 2019, the Constitutional Court ruled the criminal offense of abortion (termination of pregnancy) unconstitutional, citing respect for women's right to bodily self-determination. As of January 1, 2021, abortion was effectively legalized. The Constitutional Court ordered the National Assembly to come up with alternative legislation to reflect the decision, but 4 years have passed without such legislation being enacted. In the 21st National Assembly, 7 bills to abolish the abortion law were introduced, including amendments to the Criminal Code and the Mother and Child Health Act, but were never properly discussed until the end of the 21st NA period.
- Company
- Competition rises in 1st-line urothelial cell carcinoma mkt
- by Eo, Yun-Ho Jul 31, 2024 05:51am
- The battle for the first-line treatment market for urothelial cell carcinoma (bladder cancer) is heating up amongst anticancer drugs. In addition to the PD-L1 immuno-oncology drug Bavencio (avelumab), which is already being reimbursed in Korea, the PD-1 immuno-oncology drug Opdivo (nivolumab) in combination with cisplatin and gemcitabine, and the antibody-drug conjugate (ADC) Padcev (enfortumab vedotin) in combination with the PD-1 immuno-oncology drug Keytruda (pembrolizumab) have entered the market one after another. The Opdivo and the ‘Padcev+Keytruda’ combination have garnered particular attention due to their recent almost simultaneous indication expansions. It remains to be seen whether these drugs will be successful in gaining reimbursement in Korea. The approval of Opdivo was based on the results of the Phase III CheckMate-901 study, which compared patients receiving either nivolumab in combination with cisplatin and gemcitabine (up to 6 cycles) followed by nivolumab alone for up to two years or cisplatin and gemcitabine (up to 6 cycles). Key results showed a statistically significant improvement in the study’s primary efficacy endpoint, overall survival (OS), and progression-free survival (PFS) as assessed by a blinded independent central review committee (BICR) in the Opdivo combination arm. The safety profile of the Opdivo regimen observed in the study was consistent with previously reported studies, with no new safety signals identified. The efficacy of the Padcev+Keytruda combination was demonstrated in the Phase III EV-302/KEYNOTE-A39 study. In the study, the primary endpoint, median progression-free survival (PFS) was 12.5 months in the Padcev+Keytruda arm, significantly higher compared with the 6.3 months in the chemotherapy arm. The secondary endpoint, median overall survival (OS), was 31.5 months, nearly doubling the 16.1 months observed in the control arm. In August 2023, Bavencio became the first immuno-oncology drug to receive reimbursement in Korea as a first-line maintenance treatment in adult patients with locally advanced or metastatic urothelial cell carcinoma whose disease has not progressed with first-line platinum-based chemotherapy.
- Company
- Clinical trials for K-pharma's new lung fibrosis drugs
- by Son, Hyung-Min Jul 31, 2024 05:51am
- Pharmaceutical and biotech companies in South Korea are developing new drugs for Idiopathic pulmonary fibrosis (IPF). BridgeBio's Bersiporocin has progressed to Phase 2 trials, and the company has recently completed registering patients for Phase 2 trials. Nextgen Bioscience received Phase 1 investigational new drug (IND) approval from the Ministry of Food and Drug Safety (MFDS) earlier this month and will begin assessing the potential of commercialization. Daewoong PharmaceuticalAccording to industry sources on July 31st, Daewoong Pharmaceutical received a recommendation from the second independent data monitoring committee (IDMC) meeting to continue clinical trials. At the first IDMC meeting in March and the second meeting, Bersiporocin's safety data presented no issues. IDMC comprises experts who independently monitor the safety of study participants and pharmaceutical efficacy during clinical stages. As an independent committee with objectivity, IDMC recommends sponsors regarding the trial's progress, study participant recruitment delays, clinical design changes, and discontinuance of clinical trials. Bersiporocin is a new drug candidate and a PRS inhibitor that provides anti-fibrotic effects to collagen synthesis. Bersiporocin's mechanism involves inhibiting the excessive synthesis of collagen, known as the cause of fibrosis. IPF is a form of interstitial pneumonia characterized by progressing pulmonary fibrosis. This disease has unmet needs because currently available treatments have limited treatment effects. Bersiporocin was evaluated for its safety and pharmacokinetic properties, including absorption, distribution, and metabolism, in a Phase 1 clinical trial involving 162 healthy adults in South Korea and Australia in 2022. The following year, Daewoong Pharmaceutical received approval for a multinational Phase 2 trial to assess the efficacy and safety of Bersiporocin in patients with idiopathic pulmonary fibrosis aged 40 and older. Currently, this trial involves patients either on approved treatments or have discontinued them. The Phase 2 trial is being conducted in 10 institutes in South Korea, including Asan Medical Center and Severance Hospital, and about 20 institutes in the United States. To date, this trial recruited 61 patients, and the total patient cap is 102. BridgeBio completes registering patients for Phase 2 trial…Nextgen enters clinical trial In addition to Daewoong Pharmaceutical, BridgeBio, Nextgen Bioscience, and iLeadBMS, Ildong Pharmaceutical's subsidiary, are conducting clinical trials for new drugs to treat IPF. BridgeBio has recently completed registering 120 patients for the Phase 2 trial of its new drug candidate, ‘BBT-877,’ for IPF. The Phase 2 trial is evaluating the efficacy, safety, and tolerability of BBT-877 compared to placebo in 120 patients with IPF. It is now being conducted in 50 institutes across South Korea, the United States, Australia, Poland, and Israel. BBT-877 is an innovative new drug candidate, selectively inhibiting the new targeted protein, autotaxin. Autotaxin is a protein known to bind receptors and is involved in pathologies of sclerosis and tumorigenesis. BridgeBio secured an exclusive global license of BBT-877 from LegoChem Biosciences (currently LigaChem Biosciences) in 2017. In May, BridgeBio received a recommendation from IDMC to continue its clinical trial. Based on data evaluating the efficacy and safety of BBT-877 in 75 study participants, no issue has been found regarding the drug's safety and effects. Earlier this month, Nextgen Bioscience received IND approval from the MFDS for its Phase 1 clinical trial of a new drug candidate, ‘NXC680.’ Designed to work by an anti-fibrotic mechanism, NXC680 selectively inhibits autotaxin, which is known to be involved in various fibrotic diseases. In January 2023, NXC680 received an Orphan Drug Designation from the U.S. Food and Drug Administration (FDA). Ildong Pharmaceutical's subsidiary, iLeadBMS, is developing new drugs in addition to Pirespa (pirfenidone), which can delay IPF symptoms. iLeadBMS's IL1512 works by targeting CXCR7, which promotes inflammation and fibrosis. Through this mechanism, IL1512 regulates the mechanism associated with the progression of liver fibrosis, such as fibroblast activation, organ recovery, and angiogenesis. In a preclinical study, IL1512 demonstrated improvements in bleomycin-induced skin fibrosis mouse model. Ildong Pharmaceutical is developing a generic medicine referencing Ofev (Nintedanib), previously released by Boehringer Ingelheim. Ofev is a targeted therapy with proven effects to delay IPF disease progression. Ofev data indicate that it can reduce lung function reduction by 50%. Il Dong Pharmaceutical has recently completed the biological equivalence testing of Ofev generic and is analyzing the data. Ildong Pharmaceutical plans to target the IPF market with Pirespa and its new drug, Ofev generic.
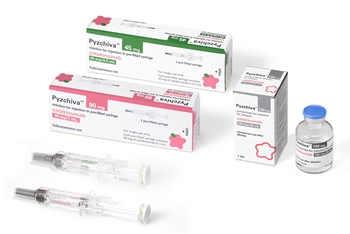
- Company
- Samsung Bioepis launches Stelara biosimilar Pyzchiva in EU
- by Hwang, Byung-woo Jul 30, 2024 05:52am
- Samsung Bioepis has launched Pyzchiva, its biosimilar version of Stelara (ustekinumab), in the European market. Pic of PyzchivaPyzchiva was approved in Europe and Korea in April and was launched into the Korean market as Epyztek. The drug was approved in the U.S. in June. The company’s marketing partner, Sandoz, will take charge of its sales in Europe. Samsung Bioepis signed a partnership agreement with Sandoz in September last year to sell Pyzchiva in North America and Europe Stelara is a treatment for autoimmune diseases such as plaque psoriasis, psoriatic arthritis, Crohn's disease, and ulcerative colitis that was developed by Janssen. It posts annual global sales of approximately KRW 14 trillion (USD 10.858 billion). With the launch of Pyzchiva, Samsung Bioepis has now launched 8 biosimilars in Europe. In addition to the 3 tumor necrosis factor-alpha (TNF-α) inhibitors, Samsung Bioepis has expanded its autoimmune disease treatment portfolio in Europe by adding an interleukin inhibitor. "Our goal is to ensure that patients across Europe have access to life-changing medicines, and Pyzchiva marks an important milestone as one of the first ustekinumab biosimilars released in Europe," said Rebecca Guntern, Region President of Sandoz Europe. Meanwhile, Samsung Bioepis has been directly selling Epyztek in Korea. The company’s strategy was to strengthen its marketing capabilities and increase profitability by adding Epyztek to the list of autoimmune disease treatments it has been selling directly since March. According to the 'Drug Reimbursement List and Ceiling Price Table' as of July 1 by the Health Insurance Review and Assessment Service, Epyztek’s drug price was set at KRW 1,298,290 for the 45mg/0.5ml prefilled Inj. This is about 40% lower than the price of the original drug. Stelara’s price is also set to be reduced following the launch of the biosimilar, but Epyztek offers a price benefit even with the original drug’s price discount.