- LOGIN
- MemberShip
- 2026-05-04 12:43:52
- Company
- K-pharma speeds up Keytruda biosimilar development...
- by Kim, Jin-Gu Jun 19, 2024 05:46am
- Product photo of Keytruda. Biopharmaceutical companies in South Korea are fast-developing biosimilars referencing 'Keytruda (pembrolizumab),' which has ranked no.1 in global sales. Samsung Bioepis and Celltrion have initiated global phase 3 trials. Due to Keytruda’s leading position in global sales, competition is expected among the companies mentioned above, as well as U.S.-based Amgen and Swiss Sandoz, to develop biosimilars. Pharmaceutical companies in Korea and overseas are initiating global phase 3 trials for 'Keytruda biosimilar' According to industry sources on June 17th, Celltrion recently submitted an IND to the U.S. Food and Drug Administration (FDA) for its 'CT-P51,' which is being developed as a biosimilar referencing Keytruda. Celltrion will conduct a comparative effectiveness study to demonstrate equivalence between Keytruda, the original pharmaceutical, and CT-P51 in 606 patients with non-small cell lung cancer (NSCLC). Previously, Samsung Bioepis initiated a global Phase 3 trial for Keytruda biosimilar, 'SB27,' in April. Samsung Bioepis plans to conduct a phase 3 trial comparing the effectiveness and safety of SB27 to Keytruda, enrolling 616 patients with metastatic NSCLC across 14 countries. Samsung Bioepis (left), Celltrion. The analysis shows that competition to develop Keytruda biosimilars among companies, including biopharmaceutical companies in South Korea, globally is on the rise. Currently, U.S.-based Amgen and Swiss-based Sandoz have commenced phase 3 global clinical trials to develop Keytruda biosimilars. Amgen commenced a phase 3 global clinical trial for ABP234 in April of last year, and Sandoz initiated a phase 3 clinical trial for GME751 the same month. Keytruda’s substance patent expires after 2029…biosimilar companies aim for 'agreement' with the original company Global companies are expected to complete their biosimilar development between 2025 and 2026. Keytruda’s substance patent will expire in November 2029 in the United States and in January 2031 in Europe. Other patents, such as the use patent, formulation patent, therapeutic method-related patent, and process-related patent, tied to indications are still valid. Since Keytruda has over 30 approved indications, more than tens of patents are tied to these. The original company, with an 'evergreening' strategy, has secured the patent validity of Keytruda until after 2040 through numerous patents. Since there are more than tens of patents to overcome, companies developing biosimilars aim to reach an 'agreement' with the original company rather than launching products after patent challenges. Previously, in the cases of global pharmaceuticals, such as Humira and Enbrel, companies developing biosimilars launched their products through agreements with the original company, either by paying royalties or adjusting release dates. For these reasons, pharmaceutical industry experts anticipate that the timing of completing clinical trials will be a crucial variable for biosimilar developers. Completing biosimilar development quickly can provide an advantageous position over competing firms in negotiations over royalty payments to original companies or adjustments to launch dates. Top 10 pharmaceuticals in global sales prospects for 2024 (source: Evaluate) According to the pharmaceutical market research firm Evaluate, Keytruda generated US$25 billion (approximately KRW 33 trillion) in global sales last year, becoming the top-selling pharmaceutical globally. Previously, the top-selling pharmaceuticals were Humira from AbbVie from 2019 to 2020, and Pfizer's COVID-19 vaccine Comirnaty from 2021 to 2022. Keytruda is rapidly expanding sales through actively adding indications. After reaching US$10 billion (approximately KRW 13.3 trillion) in 2019, it showed steady growth topping US$20 billion (approximately KRW 26.7 trillion) in 2022. To date, Keytruda has secured more than 40 indications. In the Korean market, Keytruda is also a leading pharmaceutical with the highest sales. According to the pharmaceutical market research firm IQVIA, Keytruda generated sales of KRW 398.7 billion last year, up 66% from KRW 239.6 billion in 2022.
- Company
- Suhee Shin appointed new General Manager of Amgen Korea
- by Eo, Yun-Ho Jun 19, 2024 05:46am
- Suhee Shin, new General Manager of Amgen Korea Suhee Shin, the former head of Novartis Korea, is coming back to head another Korean subsidiary of a multinational pharmaceutical company after two and a half years. According to industry sources, Director Suhee Shin, who had been leading the Healthcare Innovation Cluster at Roche Korea, has been appointed to take over as the General Manager of Amgen Korea following the retirement of the current GM, Sang-kyung Noh. Shin resigned in 2022 when Novartis Korea's specialty drug and anticancer business units were merged as one and the current president, Byung-Jae Yoo took over as head of the new combined Novartis Korea. Since then, Shin joined Roche in February 2023. She left Roche last week after accepting the General Manager role at Amgen Korea. Since joining Handok Pharmaceuticals in 1999, Shin and has held commercial business unit leadership roles in various chronic disease areas including diabetes at Sanofi Korea and AstraZeneca Korea. In 2018, Shin served as the Head of the Hematology Business Franchise of Novartis Oncology and was appointed as the General Manager of Novartis Oncology in 2019, where she led the successful launch and reimbursement of various innovative oncology pipelines. Shin graduated from Ewha Womans University College of Pharmacy and earned an MBA from New York University Stern School of Business.
- Company
- New multiple sclerosis drug Ocrevus is approved in Korea
- by Son, Hyung-Min Jun 19, 2024 05:46am
- Dr. Ho Jin Kim, Professor of Neurology, the National Cancer Center A new drug for multiple sclerosis (MS), an intractable disease characterized by a high relapse rate, has been introduced to the market. Roche's Ocrevus was approved in Korea for the treatment of relapsing-remitting MS as well as primary progressive MS, for which there had been no treatment options available. Medical experts have expressed the importance of starting treatment with a high-potency agent early in the disease to prevent relapses. On the 18th, Roche Korea held a press conference at the Lotte Hotel in Jung-gu, Seoul, to celebrate the approval of Ocrevus (ocrelizumab) as a treatment for multiple sclerosis in Korea. Ocrevus was approved in Korea on March 13 as an autoimmune disease treatment that targets CD20-expressing B cells that affect the demyelinating process that causes neurological disorders in MS patients. With the approval, Ocrevus is available for the treatment of Relapsing forms of MS, to include clinically isolated syndrome, relapsing-remitting disease, and active secondary progressive disease, in adults, and primary progressive MS, in adults. MS is a chronic disease in which myelin sheaths are damaged by an autoimmune inflammatory response. Damage to the myelin sheath causes symptoms such as muscle weakness, fatigue, and vision impairment, and can lead to non-traumatic neurological disability. As of 2022, an estimated 2,674 people in Korea are living with MS, with more than 62% of them in the 20-40 age group. Until now, antibody drugs such as Tysabri (natalizumab), Gilenya (fingolimod), and MabThera (rituximab) were used for the disease, but there had been a steady need for additional high-potency drugs. Various new drugs have been developed overseas, including Novartis' Kesimpta (ofatumumab) and TG Therapeutics' Briumvi (ublituximab), but Roche's Ocrevus is the only one introduced in Korea. Ocrevus also offers greater dosing convenience over Kesimpta (administered once a month), as it can be administered every 6 months. The approval was based on the Phase III OPERA-I and II trials. The trials evaluated the efficacy and safety of Ocrevus versus Biogen's interferon therapy Plegridy (peginterferon beta-1a) in patients with relapsing-remitting MS. In the trials, Ocrevus reduced the annualized relapse rate (ARR) by nearly half compared to Plegridy. Specifically, in the OPERA I trial, the ARR was 0.156 in the 96-week Ocrevus arm and 0.292 in the control arm, and in the OPERA II trial, the ARR was 0.155 in the 96-week Ocrevus arm and 0.290 in the control arm. Ocrevus also showed efficacy in the Phase III ORATIORIO trial that studied patients with primary progressive MS. In this study, Ocrevus reduced confirmed disability progression (CDP) by 24% over 12 weeks compared to the control arm. In terms of safety, the most common adverse events were infusion-related reactions (IRRs) and upper respiratory tract infections, most of which were mild to moderate in severity. Dr. Ho Jin Kim, Professor of Neurology at the National Cancer Center, said, "The unmet need for MS treatments remains high. In Korea, there is no opportunity to use high-potency drugs in the treatment of MS, and we have low access to such drugs. Overseas, high-potency drugs have been used as first-line treatment for MS since 2020. Ocrevus was approved overseas in 2017, but it took 7 years for it to be approved in Korea." He added, “In MS, even the smallest differences in its early stages can have enormous cumulative effects. This is why there are significant benefits to having early access to highly effective treatments. These treatments will not only improve the quality of life for the patients but will also help reduce socioeconomic burden. Ocrevus will be well utilized because it owns ample data on its efficacy as well as on long-term administration.”

- Company
- "Novotech is a global CRO partner for Korean biotech"
- by Lee, Tak-Sun Jun 19, 2024 05:46am
- Novotech is a global full-service clinical Contact Research Organization (CRO) focused on partnering with biotech companies to accelerate the development of innovative and novel therapies at every clinical phase. Recognized for its CRO industry-leading contributions, Novotech received numerous prestigious awards, including the CRO Leadership Award 2023, Best Cell & Gene Therapy CRO 2023, and Asia-Pacific Contract Research Organization Company of the Year Award 2023, since 2006. Novotech was founded in 1997 and provides comprehensive services, including a research center, phase 1 clinical trial facility, drug development consulting, and regulatory expertise. The company has experience in over 5000 clinical projects, including Phase 1 to Phase 4 clinical trials and bioequivalence tests. Furthermore, Novotech has 34 offices globally and has over 3000 staff. The company partners with over 1500 CRO firms, establishing itself as a partner with an end-to-end strategy. Commencing in Asia Pacific, which is one of the regions with the most complicated clinical trials, Novotech has strategically expanded its geological position with increased customer needs for global clinical trials. In 2008, Novotech opened a subsidiary in South Korea, one of the leading global countries in clinical trials. The decision was based on South Korea’s continuous investment in research and development, rapidly growing with the government support, and focusing on life sciences and biotechnology. In 2020, Novotech acquired PPC, which has subsidiaries in Taiwan, China, and South Korea. Through this acquisition, Novotech was able to fulfill the growing biotech needs, particularly in China. In 2022 and 2023, Novotech acquired NCGS (May 2022), EastHORN (November 2023), and CBR International (January 2023). The company entered the United States and European markets, strengthening its global position. Sanghee Kim (53), Korea Country Managing Director, is leading Novotech Korea, which is growing as a biotech hub. Kim has led the growth of PPC in South Korea through integration with Biosuntek Laboratory, which was one of the CROs in South Korea, and later PPC integrated with Novotech. "Previously, Novotech provided multinational clinical trial services, and PPC focused on phase 1 to phase 4 trials in South Korea and PMS. With the acquisition of Biosuntek, which specializes in bioequivalence tests and PK analysis, Novotech now offers the most comprehensive service among global and domestic CROs," Kim said. Sanghee Kim, Novotech Korea The Korea subsidiary of Novotech focuses on novel drug development consulting, protocol development, project management and clinical monitoring, pharmacovigilance, data management (DM), and statistical analysis. Furthermore, with people specializing in areas such as site management organization (SMO), the company can offer full-service for domestic and global clinical trials. "Most global CROs do not have people specializing in DM/statistical analysis and medical, Lab, and SMO in South Korea," Kim said. "Novotech Korea has advantages and can be differentiated from other countries, with a specialty workforce offering optimized comprehensive service to fulfill the needs of biotech and pharmaceutical companies in all phases, including the early and late phase clinical trials," Kim explained. After graduating from the College of Pharmacy at Chung-Ang University, Kim has been working in the pharmaceutical and CRO industry since 1994. She started her career in a pharmaceutical company in Korea, focusing on approval applications and academic training, and relocated to a multinational pharmaceutical company, taking on various roles related to clinical trials in the medical department. "While working in the pharmaceutical industry, I had interest in various businesses and took on the roles. I have gained valuable experiences working for 15 years in clinical trial planning and management, pharmacovigilance, and quality management," Kim said. As many pharmaceutcial companies considered clinical trial outsourcing as the key strategy in mid-2000, Kim beame interested in CRO and started her CRO career at PPC Korea with a passion for working in a new field. "PPC was a small company, less than a year since commencement. As the first country manager in the Korean subsidiary, I was involved in setting up the departments, and the company gradually grew into CRO with the full-service. In the beginning, colleagues and employers I met in my previous work helped in many ways, and it became a motivation. Afterward, I established a reputation from clients," Kim said. With a 30-year career in pharmaceuticals and CRO, Kim says fulfilling the needs of everyone involved in the business is a responsibility and an important task as a leader. "As a Country Managing Director, I value stakeholder management. Many leaders in the CRO industry value human resources management as a key task. I believe, like other service businesses, that CRO operations ultimately aim for balanced stakeholder management and management that contribute to better health outcomes for employees, customers, shareholders, local communities, and patients. If we can achieve this, it will be a win-win situation for everyone involved," Kim commented. Novotech has been making efforts to promote a lateral organization and corporate culture that respects and supports each member. As part of our ongoing efforts to establish this corporate culture, we are honored to be recognized as one of the top 100 companies to work for in South Korea in 2022 (Great Place To Work® Korea). In response to a question about the most memorable project, "Whether it's clinical trials for regulatory approval or post-market studies, all projects aim to generate the basis of efficacy, safety, and utility," Kim said, adding, "Once a project completes and receives market approval from regulatory authorities, receiving a letter of appreciation from the client is a memorable and fulfilling experience. However, because all projects are meaningful and important to our clients, it's difficult to single out just one project." "South Korea has excellent facilities and infrastructure in clinical trial institutions (hospitals) and outstanding researchers, creating an environment for active clinical research," Kim said. However, Kim also analyzed, "In recent years, the market has not seen significant growth, and competition has intensified with the entry of numerous global CROs both domestically and internationally." "Regulatory adjustments are necessary since the digital transformation of clinical trials has accelerated since COVID-19," Kim said. "Improving clinical trial regulations sometimes requires understanding and cooperation from multiple government agencies depending on the case, which hinders rapid regulatory improvements," Kim added. "We understand that the needs vary among biotech companies, large domestic pharmaceutical companies, small to medium-sized pharmaceutical companies, and multinational companies. Accordingly, we aim to be a trusted partner in every stage, from consulting on development strategies, depending on whether our clients' R&D goals are focused on obtaining domestic market approval, licensing out after initial clinical results, or conducting clinical trials for global market entry and subsequent market approvals in multiple countries," Kim concluded.
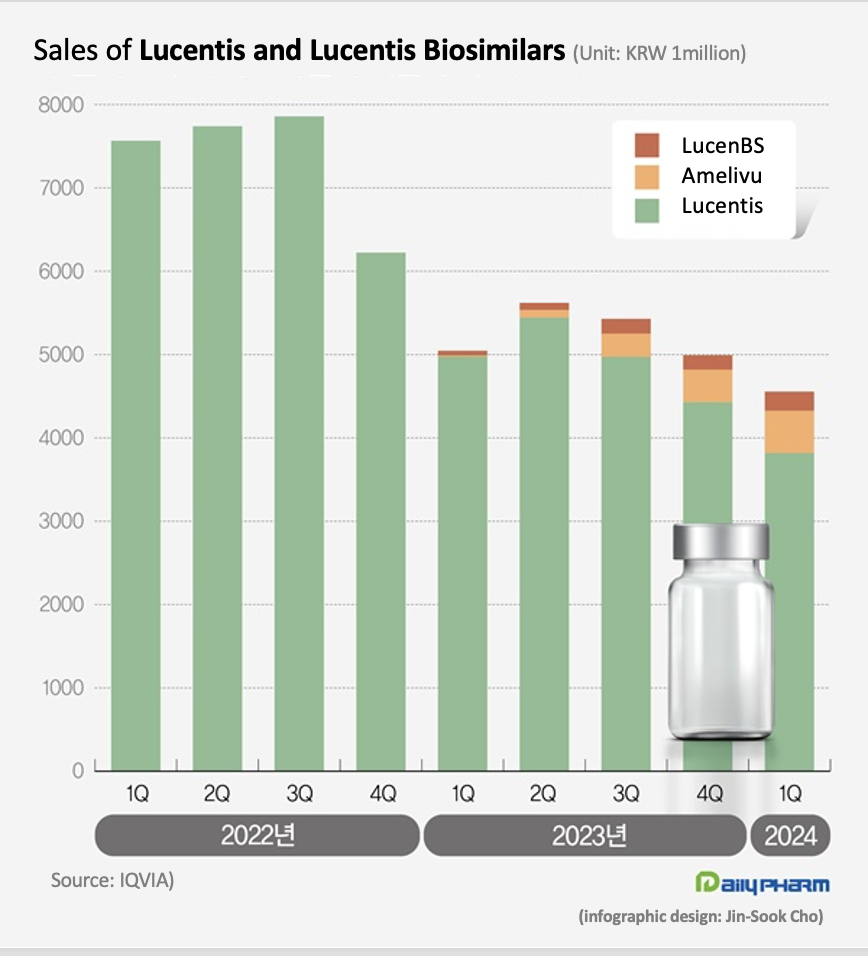
- Company
- Lucentis biosimilars occupy 16% of mkt in 1 year
- by Chon, Seung-Hyun Jun 19, 2024 05:46am
- The market size for the eye disease drug Lucentis has shrunk significantly. Last year, its biosimilars entered the market and reduced the drug price of Lucentis, reducing the market size. Although the market share of the two biosimilars remains a mere 16%, the price reduction of Lucentis saved more than KRW 10 billion in annual health insurance finances. According to drug research institution IQVIA, the market size of ranibizumab in Q1 was KRW 4.6 billion, down 9.7% YoY. Ranibizumab is the active ingredient contained in Lucentis. Lucentis, which is marketed by Roche and Novartis, is a drug used to treat eye diseases such as macular degeneration and diabetic macular edema. It is indicated for the treatment of ▲neovascular (wet) age-related macular degeneration, ▲diabetic macular edema, ▲proliferative diabetic retinopathy, ▲ vision impairment due to macular edema following retinal vein occlusion, ▲and vision impairment due to choroidal neovascularization. The ranibizumab recorded KRW 7.6 billion in sales in Q1 2022, down 33.3% from KRW 5 billion in Q1 last year. The market size for Lucentis shrank due to the drug price cut that followed the entry of its biosimilars. Samsung Bioepis and Chong Kun Dang received approvals for their Lucentis biosimilar products. Samsung Bioepis received approval for its Lucentis biosimilar Amelivu in May 2022, and Chong Kun Dang received approval for its LucenBS in October 2022. Amelivu and LucenBS have been reimbursed since January last year. The insurance price ceiling of Lucentis has been cut by 30% since February last year. The price of Lucentis 10mg (2.3mg/0.23mL) had fallen 30% from KRW 828,166 to KRW 579,716. The price of Lucentis Prefilled Syringe (KRW 826,231→KRW 578,362) also fell 30%. As a result, sales of Lucentis in Q1 were 3.8 billion won, down 23.3% YoY. Compared to Q1 2021, which was before the price cut, the company's sales have fallen by 49.5% in 2 years. While domestically developed biosimilar versions of Lucentis are working to accelerate market penetration, their current share in the market is less significant. In Q1, Amelivu sold KRW 500 million. Since its sales exceeded KRW 100 million in Q3 last year, its sales scale has gradually expanded, but its gap with the original drug is still large. Amelivu’s market share in Q1 was 11.1%. LucenBS’s Q1 sales were KRW 200 million, accounting for only 5.0% of the total market. The two biosimilars combined occupied 16.2% of the market, falling far behind Lucentis’s share. However, Amelivu and LucenBS’s supply at a relatively low price in the market is said to have significantly contributed to reducing the cost of medication for patients. The insurance ceiling price of Amelivu 10mg/ml is KRW 350,000, which is only at a 60.4% level of Lucentis’ price. Compared to the price of Lucentis before the price cut, it costs 42.3% of Lucentis’ previous price. The insurance price of LucenBS is KRW 150,000, which is 74.1% lower than Lucentis. This is more than 80% lower than the price of Lucentis before the biosimilar was launched. Initially, Chong Kun Dang listed LucenBS at an insurance price of KRW 300,000, then further reduced the price by 50% this year. In Q1, the Lucentis market shrank by 40.0% compared to 2 years ago. However, considering the drug price cut and the market entry of cheaper biosimilars, their usage has not decreased. The emergence of domestically developed biosimilars has resulted in annual health insurance savings of more than KRW 10 billion.

- Company
- Targeted therapies for HER2m NSCLC actively developed
- by Son, Hyung-Min Jun 18, 2024 05:49am
- The domestic and foreign pharmaceutical industry has taken up the challenge of developing a new drug for HER2-positive NSCLC. After Enhertu opened the door by obtaining the first marketing authorization as an anticancer drug targeting the HER2 mutation in Korea, many later entrants such as Yuhan Corp, Boehringer Ingelheim, and Hanmi Pharmaceutical have also started clinical trials. Until now, there have been a number of drugs targeting various gene mutations such as EGFR, ALK, ROS1, BRAF, MET, and RET in NSCLC, but none has targeted HER2. Approximately 2-4% of patients with NSCLC have a HER2 mutation. Due to its low prevalence, the companies had difficulty developing targeted therapies. This is why the clinical results of the latecomers are gaining interest. Boehringer Ingelheim disclosed early phase clinical trial results confirming the efficacy of its HER2-targeted antitumor candidate, and Yuhan Corp secured multinational IND approval for its HER2-mutation targeted antitumor drug candidate after Leclaza. Hanmi Pharmaceuticals also launched a new HER2-targeted NSCLC drug after failing with poziotinib. According to industry sources on the 18th, Boehringer Ingelheim's zongertinib (Development code name: BI-1810631) showed an effect in HER2-positive NSCLC. Zongertinib is a tyrosine kinase inhibitor (TKI) that binds to the HER2 mutation without inhibiting the wild-type EGFR. The Phase 1 trial, named BEAMION-Lung01, evaluated the efficacy of zongertinib in 36 patients with metastatic/advanced NSCLC who were refractory to standard of care and had HER2 mutations. The study was designed to determine if zongertinib can slow the progression of advanced NSCLC compared to the current standard of care, Keytruda plus platinum-based chemotherapy. The primary endpoint was set as an investigator-assessed overall response rate (ORR). Trial results as of July 31, 2023, showed that the ORR was 58% with zongertinib with a disease control rate (DCR) of 97%. In terms of safety, the most common treatment-related adverse events (TRAEs) were diarrhea and rash. Based on the tolerability and safety of the drug demonstrated in the Phase 1 trial, Boehringer Ingelheim plans to conduct a full efficacy evaluation of zongertinib through the Phase 2 trial. Korean companies including Hanmi and Yuhan also start developing treatments for HER2 NSCLC Yanghwa has also jumped into the development of HER2-targeted anticancer drugs. The company plans to develop a new drug for NSCLC to add on to its EGFR (HER1)-targeting anticancer drug Leclaza. The company’s candidate, YH42946, targets NSCLC patients with EGFR exon 20 mutations along with HER2 positivity. YH42946 was approved by the U.S. Food and Drug Administration (FDA) last month and was then granted to start a multinational Phase I/II clinical trial in Korea in the middle of this month. The company acquired the new anticancer drug candidate and its pipeline last year from a local biotech J Ints Bio. The Phase I/II trial initiated this time is the first trial evaluating the tolerability and safety of YH4294 in humans. In preclinical studies, YH42946 has shown anti-tumor effects against HER2 mutations and EGFR exon 20. It has also shown an effect on solid tumors such as breast and colorectal cancers. Hanmi Pharmaceutical is also out to develop new treatments after the failure of poziotinib. Its new anticancer pipeline, the ‘selective HER2 Exon20 insertion mutation inhibitor', which had not been disclosed until now, was found to have an anti-cancer effect based on its strong activity against HER2 exon20 insertion mutation and high selectivity for EGFR and has been confirmed to be a viable treatment for NSCLC. The company has a history of developing an oral treatment for HER2-mutant NSCLC. Hanmi Pharmaceutical and its U.S. partner Spectrum (now Assertio) developed poziotinib for locally advanced or metastatic NSCLC with HER2 exon 20 insertion mutations but ultimately failed to gain FDA approval. In 2022, the U.S. Oncologic Drugs Advisory Committee (ODAC) voted (9:4) that the benefits of poziotinib did not outweigh the risks, prior to the FDA's decision on whether to grant it marketing authorization. At the time, the ODAC noted that poziotinib lacks efficacy compared to Enhertu, which also targets HER2. Currently, the only targeted therapy option available for HER2-mutant NSCLC is Enhertu, developed by Daiichi Sankyo and AstraZeneca. On March 20, Enhertu’s indication was extended to the treatment of patients with unresectable or metastatic NSCLC whose tumors harbor an activating HER2 (ERBB2) mutation and who have received prior systemic therapy, including platinum-based chemotherapy. In the DESTINY-Lung02 study, Enhertu achieved a confirmed objective response rate (ORR) of 49%, complete response (CR) of 1%, and partial response (PR) of 48% as assessed by an independent centralized blinded review (BICR). Poziotinib achieved an ORR of 28% in the ZENITH20 cohort 2 study. Following the failure to receive approval, Spectrum deprioritized the development of poziotinib and reduced its R&D workforce by 75%. Therefore, it will be interesting to see if Hanmi Pharmaceutical’s newly developed HER2-targeted cancer drug will be able to go on further and receive approval.
- Company
- Eisai’s JAK inhibitor, 'Jyseleca,' has landed at hospitals
- by Eo, Yun-Ho Jun 18, 2024 05:48am
- Eisai Korea’s Eisai Korea’s 'Jyseleca,' a JAK inhibitor, is now available for prescription at general hospitals in South Korea. According to industry sources, Jyseleca, which is the fifth JAK inhibitor in South Korea, has passed the drug committee (DC) of Big 5 tertiary general hospitals, including Seoul National University, Seoul Asan Hospital, and Sinchon Severance Hospital, and national hospitals in major cities. It has expanded prescription areas after being listed for insurance reimbursement in November of last year. Jyseleca’s initial indication for reimbursement was for the treatment of rheumatoid arthritis and moderately to severely active ulcerative colitis. The reimbursement criteria are set for individuals who have had an inadequate response to conventional therapies or have no drug tolerance to each disease. For those who are over 65 years old, the criteria are set for individuals who have had an inadequate response to TNF-α inhibitors or have no drug tolerance. In July of last year, Jyseleca received conditional approval for reimbursement from the Drug Reimbursement Evaluation Committee (DREC) of the Health Insurance Review and Assessment Service (HIRA). It is indicated for the treatment of rheumatoid arthritis and ulcerative colitis. For the approval, Eisai accepted a price 90% below the actual average, exempted from the HIRA ceiling price negotiations, and quickly became reimbursable. In South Korea, JAK inhibitors, such as 'Xeljanz (tofacitinib),' 'Olumiant (baricitinib),' and 'Rinvoq (upadacitinib),' are being prescribed. It is to be watched whether Jyseleca would have a competitive advantage over these drugs. Since their launch, these drugs have been expanding indications and reimbursement criteria. Xeljanz additionally secured indications for ulcerative colitis and psoriatic arthritis, and latecomers, such as Rinvoq, are also expanding prescription areas in autoimmune diseases, including atopic dermatitis, Chron’s disease, and ankylosing spondylitis. Meanwhile, Jyseleca is a selective ATP-competitive and reversible JAK1 inhibitor. JAK1 transmits signals from a cytokine, and it is regarded as the key target for the treatment of rheumatoid arthritis. Recently launched treatments inhibit JAK2 or JAK3, depending on their mechanism. However, there are concerns that adverse reactions may occur, as two signaling pathways are involved in regulating immune cell proliferation and homeostasis. The FINCH1, FINCH2, and FINCH3 Phase 3 trials demonstrated the effectiveness of Jyseleca. In the FINCH1 trial, Jyseleca 200 mg treatment in patients with moderately to severely active rheumatoid arthritis reached ACR20 at 20 weeks more quickly despite continued treatment with MTX.
- Company
- Noh will retire from Amgen after 9 years since its inception
- by Eo, Yun-Ho Jun 17, 2024 05:46am
- Noh Sang-kyung, Amgen Korea general manager Noh Sang-kyung (61), general manger of Amgen Korea, who led Amgen’s Korean office since its inception is set to retire from the company. According to industry sources, Noh has recently confirmed his retirement from the company. He has served in this role for nine years since the company’s inception in South Korea in 2015. His successor has recently been appointed. Noh has been recognized for contributing to the stable establishment of Amgen in Korea and for delivering Amgen’s innovative products to Korean patients. Under Noh’s management, Amgen’s Korean office was able to list six launched products, including 'Prolia (denosumab),' 'Evenity (romosozumab),' 'Xgeva (denosumab),' 'Repatha (evolocumab),' 'BLINCYTO (blinatumomab),' and 'Kyprolis (carfilzomib),' in health insurance reimbursement listing. Meanwhile, Noh graduated from Sogang University with a degree from the Department of Life Sciences and worked at Lilly Korea, Roche Korea, and BMS. After working at Bayer Schering Pharma in 2007, he was appointed as the CEO of Bayer Schering Pharma Philippines office and the Head of Pharmaceuticals Business unit of Bayer Korea. In May 2015, Noh was appointed as the first general manager of Amgen’s affiliate.
- Company
- Elafibranor receives orphan drug designation in Korea
- by Eo, Yun-Ho Jun 17, 2024 05:46am
- The biliary cholangitis drug 'elafibranor' has been designated as an orphan drug in Korea. The Ministry of Food and Drug Safety (MFDS) recently announced the designation through an announcement. Elafibranor, a dual peroxisome-activated peroxisome receptor alpha/delta (PPAR α,δ) agonist that is being developed by Ipsen, received accelerated approval from the U.S. FDA for the primary biliary cholangitis indication on Nov. 10. More specifically, the drug is indicated for the treatment of adult patients with primary biliary cholangitis who have had an inadequate response to ursodeoxycholic acid (UDCA) or who cannot tolerate UDCA monotherapy due to tolerability issues. The FDA’s accelerated approval is based on data from the Phase III ELATIVE trial, which did not demonstrate improved survival or prevention of liver function decline. Ipsen is currently conducting the ELFIDENCE trial, a confirmatory clinical trial. The results of this study will determine whether the authorities will maintain the authorization. The ELATIVE trial demonstrated that elafibranor is an effective second-line treatment for patients with PBC with favorable benefit and risk data. Meanwhile, at the European Association for the Study of the Liver (EASL) 2024 Annual Congress, 2 additional analyses from the Phase 3 ELATIVE study, which evaluated the safety and efficacy of elafibranor in 161 primary biliary cholangitis patients with inadequate response or intolerance to ursodeoxycholic acid (UDCA), were presented one after another. The presented results were from Week 72 analysis, which showed that 30 of 108 patients (28%) in the elafibranor arm and 13 of 53 patients (25%) in the placebo arm remained on treatment through Week 72. Among these patients, 70% achieved biochemical response in the elafibranor arm, whereas no patients achieved biochemical response in the placebo arm.
- Company
- Pfizer immediately reapplies to extend reimb for Lorviqua
- by Eo, Yun-Ho Jun 14, 2024 05:47am
- Pfizer Korea has quickly reapplied for reimbursement of Lorviqua, its third-generation ALK anticancer drug whose reimbursement review process recently broke down at the drug pricing negotiation stage. According to Dailypharm’s coverage, Pfizer Korea submitted an application to expand insurance reimbursement for the ALK-positive non-small-cell lung cancer (NSCLC) treatment Lorviqua (lorlatinib) to the first-line treatment on the 12th. The drug's drug pricing negotiations with the National Health Insurance Service broke down in late May. It remains to be seen whether Lorviqua will be able to secure reimbursement in its second attempt and how quickly the discussions will move forward. In January, Pfizer submitted an application to convert the drug’s reimbursement status to general listing. At the time, its reimbursement expansion process was already underway, and given that Lorviqua has already undergone a pharmacoeconomic evaluation in the first-line setting and has completed the Health Insurance Review and Assessment Service review, the drug’s conversion to general reimbursement listing status is expected to be discussed in conjunction with the first-line reimbursement expansion Regardless of whether reimbursement is expanded or not, the pharmaceutical company’s will and the government’s flexible administration will be required to achieve a quick result. Lorviqua was specifically designed to penetrate the blood brain barrier (BBB). The drug’s high clinical value as a first-line treatment was recognized in the 5-year long-term follow-up results of the CROWN study that was presented at ASCO. Study results showed that Lorviqua reduced the risk of disease progression or death by 81% compared to crizotinib, with 60% of patients surviving without disease progression at 5 years. The risk of brain metastasis progression was reduced in 94% of patients, with only 4 of 114 Lorviqua-treated patients without brain metastases developing brain metastases. The reason the drug pricing negotiations broke down the last time is believed to be related to the ‘expenditure cap amount’ rather than 'drug price'. Lorviqua was granted pharmacoeconomic evaluation exemptions when it was first listed for reimbursement. PE exemption drugs are required to be reimbursed through the RSA Expenditure Cap type scheme. As such, a new cap amount would have been derived to account for the increased usage due to the expanded reimbursement during negotiations, which Pfizer was likely unable to accept. Press conference in front of the main gate of the National Assembly on the 13th