- LOGIN
- MemberShip
- 2026-06-10 18:16:28

- Policy
- Generic market for anti-cancer drug Xtandi heats up
- by Lee, Tak-Sun May 20, 2026 02:28pm
- Original Xtandi TabThe generic market for Xtandi (enzalutamide, Astellas), a blockbuster prostate cancer treatment, is in turmoil.With the expiration of the substance patent approaching this June and approvals for soft capsule generics coming one after another, latecomers are now setting their sights squarely on the ‘tablet’ market—the original manufacturer’s latest strategic weapon— rapidly expanding the competitive battlefield.According to industry sources, on April 24, applications for approval of three film-coated tablet strengths of enzalutamide (40mg, 80mg, and 160mg) were simultaneously submitted to the Ministry of Food and Drug Safety. Industry attention is focused on these applications because they aim to secure ‘first generic exclusivity,’ considered the key to early market dominance.From capsules to tablets…follow-up approvals accelerate after breaking the 2033 barrierOriginally, the Xtandi generic market had been developing primarily around soft capsule formulations timed with the expiration of the substance patent on June 27. Major pharmaceutical companies, including Alvogen Korea, Daewon Pharmaceutical, Hanmi Pharmaceutical, GL Pharma, and Dongkook Pharmaceutical, have already obtained approvals for soft capsule generics and are preparing to launch.In response, original manufacturer Astellas Pharma Korea introduced ‘Xtandi Tab’ (40mg and 80mg) with improved dosing convenience and completed reimbursement listing last April. This was part of a defensive strategy to shift the market’s center of gravity toward tablets and fend off the generic competition. This was because the tablet formulation was protected by a separate composition patent valid until September 2033.However, the defensive line collapsed as domestic pharmaceutical companies successively succeeded in circumventing this 2033 formulation patent through a series of negative scope confirmation trials. The latest film-coated tablet generic applications are therefore interpreted as the first step toward realizing a ‘dual competition’ structure between dosage forms after neutralizing the patent barrier.Addition of a 160mg high-dose option… Aiming for exclusivity through first generic approval rightsA notable feature of the film-coated tablet lineup applied for this time is the inclusion of a 160mg high-dose product, which is not available in the original Xtandi Tab lineup (40mg, 80mg).Currently, patients face the inconvenience of having to take four 40mg soft capsules at once each day. In response to the original manufacturer introducing an 80mg tablet form to reduce the number of pills taken to two, the generic manufacturer has launched a counterattack by developing a 160mg ultra-high-dose formulation that requires taking “just one pill a day.” This is a strategy to surpass the original in terms of convenience.Furthermore, if these companies secure first generic exclusivity based on the latest applications, they will gain the privilege of blocking other tablet generics from entering the market for a certain period while competing exclusively against the original product.A pharmaceutical industry official predicted, “If the ‘first war’ takes place in June when soft capsule generics flood the market immediately after the substance patent expires, then approvals and reimbursement listings for tablet generics pursuing first generic exclusivity will ignite the ‘second war’ in the second half of the year.”As a direct confrontation approaches between the original company’s market-defense strategy through tablet conversion and domestic pharmaceutical firms seeking early dominance of the tablet market through patent circumvention and independent high-dose lineups, the industry’s attention is focused on who will take the lead in the enzalutamide market. Xtandi recorded domestic sales of KRW 38 billion last year, according to UBIST data.

- Policy
- MFDS to maintain small-packaging drug regulations
- by Lee, Tak-Sun May 20, 2026 02:28pm
- The Ministry of Food and Drug Safety has recommended that pharmaceutical companies voluntarily adjust their own packaging practices rather than granting the industry’s request to relax small-packaging regulations due to stable drug supply concerns arising from the Middle East conflict. The move is interpreted as maintaining the small-packaging regulations while expanding recognition of exceptional cases. Industry requests for a grace period on administrative penalties this year appear to have been rejected following opposition from pharmacy organizations and related groups.This is interpreted as the government’s intention to expand the recognition of exceptions while maintaining compliance with the small-packaging regulations. It is analyzed that the industry’s initial request for a suspension of administrative penalties this year was rejected due to opposition from groups such as the Korean Pharmaceutical Association.According to industry sources on the 18th, the MFDS recently sent an official notice to pharmaceutical associations requesting that each pharmaceutical company voluntarily adjust small-package supply ratios.Under the current “Regulations on Supply of Pharmaceuticals in Small Packaging Units,” pharmaceutical manufacturers and importers are required to supply at least 10% of their annual production or import volume to pharmacies in small packaging units.The standards are set at 30 tablets or fewer for bottle packaging, 100 tablets or fewer for blister packs, and 500 mL or less for syrups. Violations result in administrative penalties such as suspension of sales operations.The mandatory small-packaging rule was originally introduced to reduce excessive inventory at pharmacies and the resulting social costs from the disposal of unused stock.Recently, the pharmaceutical industry requested relaxation of the small-packaging regulations due to shortages of pharmaceutical packaging materials caused by instability in naphtha supply stemming from the Middle East conflict. In particular, the industry requested suspension of administrative penalties this year, even if mandatory small-package production ratios were not met.On the 2nd of last month, MFDS Minister Yu-Kyoung Oh visited JW Pharmaceutical to inspect IV solution packaging and announced that the ministry would swiftly pursue proactive administrative measures, including the relaxation of the small-packaging obligations.Following this, the MFDS working-level department reviewed the potential easing of the small-packaging requirement. However, pharmacy organizations, including the Korean Pharmaceutical Association, opposed such measures, citing concerns such as inventory disposal, making policy decisions difficult.Ultimately, it appears the MFDS has decided to adhere to the regulations while actively utilizing exceptions.In an official notice, the MFDS stated, “The MFDS has recommended that pharmaceutical companies voluntarily reduce the use of drug packaging materials, such as by adjusting the ratio of small-volume packaging.”The ministry added, “While complying with the minimum standards for small-package supply in consideration of the purpose and intent of the ‘Regulations on Supply of Pharmaceuticals in Small Packaging Units,’ if compliance becomes difficult due to shortages in packaging material raw supplies caused by wartime conditions, companies may submit statements on product-specific explanations and supporting materials to the MFDS under Article 5 of the regulation to receive exemption from the small-package requirements.”The measure is ultimately interpreted as maintaining the small-package supply obligation while broadly recognizing exception products.A pharmaceutical industry official stated, “In response to opposition from the Korean Pharmaceutical Association and others, it appears that the request for a suspension of administrative penalties requested by the industry has not been accepted. However, the MFDS’s request for voluntary adjustment by pharmaceutical companies can be interpreted as an intention to actively recognize exceptions.”
- Policy
- SK plasma gains approval for Revolade generic
- by Lee, Tak-Sun May 20, 2026 02:28pm
- Original drug, Novartis’ ‘Revolade’SK Plasma has officially entered the generic market for Novartis’ rare disease treatment Revolade (eltrombopag olamine). Following Pharmbio Korea, which first entered the market, SK Plasma is joining as a latecomer, turning the competition among domestic generic manufacturers into a two-way race.On the 18th, the Ministry of Food and Drug Safety approved two dosage strengths (25mg and 50mg) of SK Plasma’s thrombocytopenia and severe aplastic anemia treatment, “Revolpag Tab.”The original drug for Revolpag is Novartis Korea’s ‘Revolade.’ The indications SK Plasma received approval for this time include all key efficacy and indication areas held by the original drug.The three approved indications include: ▲Treatment of thrombocytopenia in chronic immune (idiopathic) thrombocytopenia patients who showed insufficient response to corticosteroids or immunoglobulins; ▲ Treatment of thrombocytopenia to initiate and maintain interferon-based therapy in chronic hepatitis C patients, and as ▲First-line treatment of severe aplastic anemia in pediatric patients aged 2 years and older and adults in combination with immunosuppressive therapy, as well as treatment of severe aplastic anemia unresponsive to prior therapies.However, for chronic immune thrombocytopenia and chronic hepatitis C treatment, the drug is approved only for use in clinical conditions associated with increased bleeding risk and cannot be used for the purpose of normalizing platelet counts.Previously, SK Plasma successfully circumvented three “new pharmaceutical composition” patents that had served as the core barrier protecting Revolade, winning a negative scope confirmation trial after a dispute that went all the way to the Supreme Court. This approval was achieved after fully resolving patent risks.As a result, the domestic eltrombopag olamine market is expected to enter an intense competitive phase between Pharmbio Korea’s first-mover product “Elpag Tab” and SK Plasma’s late-entry “Revolpag Tab.” Elpag launched in October 2024.The Revolade market has continued to grow after reimbursement criteria were expanded in 2024, reaching import sales of approximately USD 5.23 million (around KRW 7.8 billion).A pharmaceutical industry official commented, “With Pharmbio Korea already in the market and expanding its market share, SK Plasma has secured approval and is leveraging its full indication coverage. Once reimbursement listing and pricing procedures are completed and the product launches in earnest, treatment options for physicians and patients will expand significantly.”
- Policy
- Darzalex SC and Omjjara complete drug price negotiations
- by Jung, Heung-Jun May 19, 2026 11:08am
- Janssen Korea’s multiple myeloma treatment, Darzalex SC (daratumumab), is expected to be listed for reimbursement after completing drug price negotiations with the National Health Insurance Service.In addition, Korea GSK’s new myelofibrosis treatment Omjjara Tab (momelotinib) 100mg, 150mg, and 200mg has also reached a pricing agreement and will enter reimbursement listing.According to industry sources on the 18th, Darzalex SC and Omjjara Tab, which entered negotiations with the NHIS in March, recently finalized pricing agreements.Both drugs are new therapies that passed the Drug Reimbursement Evaluation Committee in January. Darzalex SC was recognized as appropriate for reimbursement as “combination therapy with bortezomib, cyclophosphamide, and dexamethasone in newly diagnosed light-chain amyloidosis patients.”Unless unexpected issues arise, both drugs are expected to proceed with reimbursement listing next month.Janssen Korea is continuing to expand the approved indications for Darzalex SC. Last April, the drug additionally received approval for 3 new indications, including combination therapy with bortezomib, lenalidomide, and dexamethasone (DVRd) for newly diagnosed transplant-eligible multiple myeloma patients. Accordingly, further applications for reimbursement expansion are expected.The reimbursement appropriateness for GSK’s myelofibrosis treatment Omjjara Tab was recognized at this year’s first DREC meeting for “treatment of intermediate- or high-risk myelofibrosis with anemia in adults,” provided that the price is set below the evaluation threshold.Omjjara passed the Cancer Drug Deliberation Committee in March last year, but submission to the reimbursement evaluation committee was delayed due to issues such as the selection of comparator drugs. After approximately 10 months, it was resubmitted and cleared the first hurdle at this year’s DREC meeting.Like Darzalex SC, it entered NHIS drug price negotiations in March and ultimately reached a final agreement. Reimbursement listing is scheduled for next month.Meanwhile, negotiations for Mounjaro, which entered price negotiations in January this year, broke down. Although the company applied for the flexible pricing contract system, it is reported that the parties reportedly failed to narrow differences regarding the separately negotiated amount.

- Policy
- Multi-listing rule hits non-innovative companies harder
- by Jung, Heung-Jun May 19, 2026 11:08am
- The ‘multi-product listing management’ rule, introduced to prevent excessive proliferation of generics, will operate far more harshly against non-innovative companies.While innovative and quasi-innovative companies will receive a three-year grace period, general companies will face immediate price cuts to 30.6%-38.5%, depending on whether they pass bioequivalence testing, leading to a significant widening of the price gap.According to industry sources on the 18th, the impact of the multi-product listing penalty system, newly introduced starting this August, will differ dramatically depending on a company’s innovation classification.Impact of the multi-product listing penalty systemThe Ministry of Health and Welfare recently issued an administrative notice revising the “Standards for Drug Price Determination and Adjustment,” newly introducing a provision applying 85% of the calculated price once the total number of identical formulations reaches 14 or more. Drugs eligible for additional premiums will also be subject to the 85% rule once the premium period ends.For innovative and quasi-innovative companies, the drug price premium period is 1+3 years if domestic manufacturing conditions are met. In other words, a 3-year grace period applies to the multi-product listing penalty as well.In contrast, general companies face an immediate reduction from the standard calculation rate of 45% to 38.25% once the number of listed products exceeds 14. If they failed to conduct their own bioequivalence testing, the price is reduced by an additional 20%, falling to 30.6%.For example, if a generic drug with an original product price of KRW 1,000 reaches 14 or more listed generics, an innovative company would maintain a price of KRW 600 won, reflecting a 60% premium, for four years before eventually dropping to KRW 382.5. A quasi-innovative company would maintain KRW 500 under a 50% premium for four years before dropping to KRW 382.5.However, the generic drug of a general company would sell at KRW 450 for only one year under the standard calculation rate before falling to KRW 382.5.This means that while innovative and quasi-innovative companies maintain prices of KRW 500–600 for three years by meeting domestic manufacturing requirements, general companies immediately face deteriorating profitability at KRW 382.5.If an ordinary company additionally failed to meet the bioequivalence testing requirement, the price would fall to KRw 306, creating nearly a twofold price gap compared with innovative companies.Ultimately, this structure ensures that the penalty for listing multiple products is applied more strictly to general companies, and a sales gap with innovative and quasi-innovative companies is inevitable during the grace period for the price adjustment.In effect, general companies effectively lose the advantage of product listing after the 14th generic entrant, subject to the multi-product pricing rule, enters the list.
- Policy
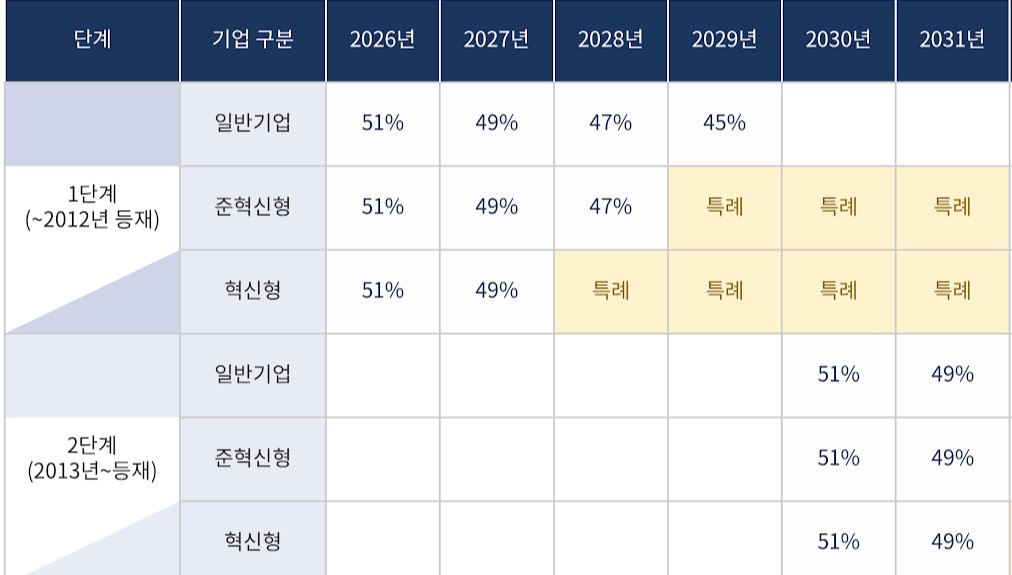
- Price cuts on existing drugs divided into two phases
- by Jung, Heung-Jun May 18, 2026 09:11am
- As the government proceeds with planned pharmaceutical pricing system reforms, follow-up discussions are expected regarding specific classification criteria for price reductions of pre-listed drugs.Key issues include how to distinguish between first-phase and second-phase price reductions for two- and three-drug combination products, as well as how and when later bioequivalence testing should be reflected in drug price reductions.According to industry sources on the 15th, specific criteria for price reductions on pre-listed drugs have not yet been finalized. Discussions on price reductions for pre-listed drugs were not properly addressed during the working-level consultative body meeting between the government and the pharmaceutical industry held in late April.Previously, the Health Insurance Review and Assessment Service (HIRA) decided to divide drugs into two groups based on their listing date in 2012. Price reductions for ingredients listed before 2012 will begin immediately within this year, while reductions for Phase 2 drugs, listed after 2012, will begin in 2030.The timing of implementation differs depending on whether a product is classified into the first or second phase of existing drug price cutsHowever, opinions remain divided regarding the classification of combination products. If the individual ingredients making up a combination drug belong to both Phase 1 and Phase 2 groups, authorities must determine at which timing the combination product itself will face price reductions.The government has stated that if even one ingredient in a combination product was listed before 2012, the combination drug would be classified as a Phase 1 drug.However, there are many issues that require consultation with the industry, such as how to handle cases where even a single ingredient retains a patent or has PMS remaining.Another key issue is how to reduce prices for drugs that failed to meet required standards due to a lack of bioequivalence testing. The reduction rate under the differentiated standards system has been increased from 15% to 20%.For example, if a product price was lowered to 45% and bioequivalence testing was not conducted, it would fall further to 36%. If a rate of 49% is applied, being a product from an innovative pharmaceutical company, it would become 39.2%, while applying a quasi-innovative rate of 47% would drop the price to 37.6%.Since price reductions for pre-listed drugs are implemented over a 10-year period, including a grace period, it is also important to determine how to reflect the results if bioequivalence testing is conducted during that period.In particular, since Phase 2 drugs will only begin facing reductions in 2030, some pharmaceutical companies may attempt to protect reduction rates by conducting bioequivalence studies.Accordingly, industry players are expected to argue that satisfying bioequivalence requirements by the final year converging toward the calculation rate should still qualify as meeting standard requirements.
- Policy
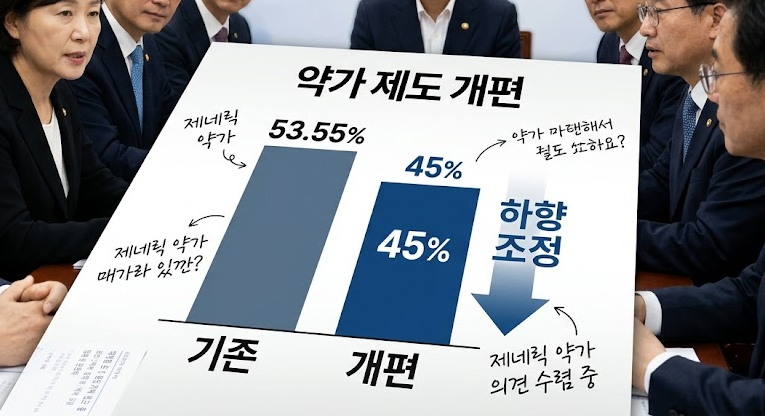
- Calculation rate for generic drug prices set at 45%
- by Lee, Jeong-Hwan May 18, 2026 09:11am
- The government has announced that a drug pricing reform plan, which cuts the drug price calculation rate for currently listed generics from 53.55% to 45%, will take effect on August 1st.The reform plan also includes improvements regarding the management of multiple-listed items, drug price calculations for transfers and acquisitions, support criteria for drugs subjected to market withdrawal, and criteria for semi-innovative pharmaceutical companies.On the 14th, the Ministry of Health and Welfare (MOHW, Minister Jung Eun-kyeong) issued a public notice for the partial amendment to the "Criteria for determination and adjustment of drugs." The ministry plans to finalize the amendment after gathering public feedback by July 13th.The implementation date for the drug pricing reform specified in the amendment notice is August 1st. The government announced that it will gather opinions until July 13th.Generic drug price calculation rate 45%....products failing to meet requirements will be priced below 36%First, the drug price calculation rate for currently listed generics will be reduced from 53.55% to 45%. The calculation rate applied to generics that fail to meet the baseline requirements will also be reduced from 85% to 80%.The baseline requirements for the drug price include whether the company conducted an independent bioequivalence test and whether it used registered drug master file (DMF) ingredients.Generics that meet all baseline requirements will be priced at 45%, those meeting some requirements will be priced at 36%, and products failing to meet any requirements will be priced at 29%.In the case of tiered pricing, the price will be cut once the number of listed items with the same formulation exceeds 13. This is a tighter restriction compared to the current threshold of 20 or more items.If the sum of the newly applied product and the number of currently listed items with the same formulation exceeds 14, the upper limit price will be fixed at 85% of the calculated amount once the price premium period ends.Innovative pharmaceutical companies, semi-innovative pharmaceutical companies, and supply-stabilizing leading pharmaceutical firms will receive preferential drug pricing. Among drugs that meet all baseline requirements, items from innovative pharmaceutical companies will receive a 60% price premium. The premium rate for items from semi-innovative pharmaceutical companies or supply-stabilizing leading pharmaceutical companies will be 50%.Definitions for semi-innovative pharmaceutical companies and supply-stabilizing leading pharmaceutical companies were also established. A supply-stabilizing leading pharmaceutical company is defined as a firm whose ratio of low-profit prevention support drugs, or the ratio of billing amounts among its listed drugs, is 20% or higher.Regarding transfers and acquisitions, the government decided to restrict the succession of existing upper limit prices for items involving a change in a manufacturer's status, excluding inheritance or mergers. Even if a generic item that maintains a high drug price is acquired, the recalculated drug price will be applied from the time of the transfer and acquisition.This regulation is designed to block back-door strategies to evade drug price cuts by purchasing items that maintain high prices.Support for drugs facing production discontinuation will be strengthened. The designation criteria for these drugs are KRW 578 for oral medications, KRW 44 per minimum unit for oral liquids, KRW 3,080 for external preparations, and KRW 5,783 for injections.A new premium clause was also created for pharmaceutical companies that have contributed to supply stabilization. The premium evaluation items include the track record of stable supply fulfillment, national essential medicines, single-listed medicines, low-priced medicines, the use of domestically produced raw ingredients, an annual billing amount of less than KRW 500 million in the previous year, treatments for statutory infectious diseases, and infectious disease crises or urgent supply shortage situations.Price-volume linkage system officiated…implemented on April 1st and October 1st of each yearThe timing of drug price cuts resulting from the price-volume linkage system and the expansion of the scope of use has been unified. The amendment specified that the ex officio adjustment of the upper limit price of drugs will be implemented on April 1st and October 1st of each year, unless there are special circumstances.In addition, a basis was established for pharmaceutical companies to refund the increased expenditure on health insurance incurred during the implementation grace period to the National Health Insurance Service if a drug price adjustment is issued at a time other than the regular implementation date.Meanwhile, the MOHW plans to implement the announced reform plan on August 1st. Regulations related to the regularization of the price-volume linkage will apply starting in January of next year (2027). The first regular drug price adjustment will take effect on April 1st, 2027.
- Policy
- Merck’s two new rare disease drugs receive GIFT designation
- by Lee, Tak-Sun May 15, 2026 02:44pm
- AI-generated imageThe Ministry of Food and Drug Safety (MFDS) is speeding up domestic approval timelines by designating two rare disease treatments from Merck as subjects for Korea’s Global Innovative products on Fast Track (GIFT) program.The MFDS announced that it designated Merck’s desmoid tumor treatment ‘Ogsiveo Tab’ and tenosynovial giant cell tumor (TGCT) treatment ‘Pimicotinib Cap’ as the 70th and 71st GIFT products, respectively. This designation is a measure to support the expedited approval of new drugs from innovative pharmaceutical companies that either have no existing treatment options or have demonstrated improved efficacy and safety.Ogsiveo Tab (nirogacestat hydrobromide), which was designated as a GIFT product on April 21, is a treatment for adult patients with desmoid tumors requiring systemic therapy. The drug suppresses tumor growth by inhibiting gamma secretase (GS) and blocking Notch signaling pathways.The drug has already received approval from the US FDA (November 2023) and the European EMA (August 2025), and was designated as an orphan drug in Korea on February 24, 2026. The MFDS selected it for expedited review based on the lack of existing treatment options.Then, on April 27, “Pimicotinib Cap (pimicotinib hydrochloride monohydrate),” a treatment for tenosynovial giant cell tumor(TGCT), was added to the GIFT list. The treatment works by selectively inhibiting the colony-stimulating factor-1 receptor (CSF-1R) to block disease progression.Pimicotinib is currently under development with FDA Fast Track and Breakthrough Therapy Designation (BTD) status in the United States, but has not yet received full approval in any global market. MFDS acknowledged the drug’s potential efficacy improvements and decided to manage it as a fast-track review product.The two products designated under GIFT will benefit from review periods shortened by approximately 25% compared to standard reviews. In addition, a rolling review of prepared materials and customized consultations with professional reviewers prior to the submission of the marketing authorization application will be provided, which is expected to significantly accelerate the timeline for the drug’s introduction in Korea.An MFDS official stated, “The exact indications and efficacy will be finalized after reviewing the submitted data. We will spare no effort in the review process to ensure that patients with intractable rare diseases can be provided with new treatment opportunities as quickly as possible.”

- Policy
- New drug review timeline cut from 295 to 240 days
- by Lee, Tak-Sun May 14, 2026 09:28am
- As the Ministry of Food and Drug Safety (MFDS) moves to shorten new drug approval review timelines from 295 days to 240 days while strengthening communication with companies. The agency will introduce face-to-face meetings between companies and reviewers prior to the application stage, as well as a “checklist” for companies to self-review their data, aiming to reduce the total approval period by nearly 2 months compared to the previous process.The MFDS announced that it has prepared a revised draft of the “New Drug Product Approval and Review Procedures (Civil Servant Guidelines),” which is currently undergoing a public comment period, and plans to fully implement it starting October 1. This revision, coming approximately one and a half years after the guidelines were established in December 2024, was pursued to maximize the predictability of new drug approvals.From post-submission to pre-submission… introduction of advance face-to-face meetingsThe most significant change is the introduction of ‘pre-NDA meetings.’ Under the original 2024 version, a dedicated team was formed and the review began within 10 days of receiving the application; however, the revised version requires the company and the MFDS to begin discussions 3 months prior to application submission.When a company requests a face-to-face meeting, a dedicated team is formed immediately, and through at least 2 meetings, any deficiencies in the data will be identified in advance. This is expected to serve as a key mechanism to accelerate the entire process by reducing the time spent on ‘requests for additional information,’ which frequently occur during the official review stage.AI-generated graphic imageIntroducing a ‘checklist’ to prevent ‘insufficient data’ at the sourceThe ‘checklist’ system, which requires companies to self-verify the completeness of their submitted data, is another key change in this amendment. Previously, the MFDS would notify companies of required corrections on a case-by-case basis after receiving the data; now, companies must complete a self-inspection using a detailed checklist starting from the face-to-face meeting stage prior to application.Products that undergo this procedure will see a significant reduction in errors or omissions in their documentation, resulting in the “preliminary review” period, conducted immediately after submission, being shortened from the previous 7 days to within 3 days.Approval timeline shortened by 55 days… “295 Days → 240 Days”Through these strengthened communication systems, the Ministry of Food and Drug Safety (MFDS) has set the target approval period for new drugs at 240 days, down from the previous 295 days. This represents a reduction of approximately 55 days compared to the original regulations.In addition, the govenrment has maximized review efficiency by codifying a ‘rolling review’ procedure for frequent exchange of opinions during the review process and by moving the GCP (Good Clinical Practice) site inspection, which previously took place after the first round of supplementary submissions, to the early stages of the review (within 60 to 120 days after submission).An MFDS official stated, “This revision goes beyond simply shortening the timeline; it institutionalizes ‘pre-submission communication’ and ‘self-assessment by companies’ to overcome the limitations identified during the operation of the original version. Once the new procedures take effect this coming October, the speed at which innovative new drugs enter the market, both domestically and internationally, will increase dramatically.”The industry’s response has also been favorable. An official from the Korea Biomedicine Industry Association said, “The introduction of the checklist will help ensure that more comprehensive data is submitted when applying for new drug approval and will likely reduce the need for supplementary data in the future. This is interpreted as a positive gesture where the MFDS and companies join hands to successfully complete the product approval process.”
- Policy
- ‘Fast reimb of Lynparza and Elahere is the solution’
- by Lee, Jeong-Hwan May 13, 2026 09:10am
- There is a growing call to improve the low survival rate of ovarian cancer patients in Korea by rapidly expanding reimbursement for AstraZeneca’s Lynparza (olaparib) and AbbVie’s Elahere (mirvetuximab soravtansine).Expanding reimbursement for ovarian cancer treatments has already been included in the government’s 2026 implementation plan under the Second Comprehensive National Health Insurance Plan, and experts have noted ample rationale for improving access.On the 12th, Professor Yoo Young Lee of Samsung Medical Center presented her view at a policy forum hosted by Representative Joo Young Lee of the Reform Party, under the theme “Improving Treatment Access and Health Insurance Coverage to Enhance Ovarian Cancer Survival.”Professor Lee described ovarian cancer as the most lethal female cancer, noting that while its incidence rate is low, the mortality rate is high once diagnosed. In fact, the incidence rate of ovarian cancer in Korea is approximately 12 cases per 100,000 people, accounting for 2.4% of all female cancers and 1.2% of all cancer cases.However, the number of patients has increased steadily over the past 20 years, rising approximately 2.4-fold from 1,353 in 1999 to 3,263 in 2022. It has been on a steep upward trajectory over the past 5 years, influenced by factors such as an aging population and changes in birth rates.Notably, the 5-year survival rate for ovarian cancer stands at 66.7%, the lowest among major female cancers. Approximately 1,465 deaths occur annually, ranking it eighth in mortality among female cancers.To improve survival rates, Professor Lee emphasized the importance of early diagnosis along with aggressive maintenance therapy.One major reason for the low survival rate is that about 70% of cases are diagnosed at advanced stages (Stage III–IV), when the cancer has already spread extensively within the abdominal cavity.The low survival rate is also influenced by the disease’s specific characteristics: early symptoms are vague, there are no effective early screening methods, and because the cancer is located deep within the abdominal cavity, it metastasizes rapidly after onset.Citing the results of clinical trials (SOLO1, PRIME, PRIMA, PAOLA-1), Professor Lee stated that administering PARP inhibitors to ovarian cancer patients who test positive for HRD (homologous recombination deficiency) through genetic testing can reduce the risk of recurrence by 40–70% and mortality risk by 30–40%.Professor Lee’s point is that survival rates can be significantly improved by combining bevacizumab and Lynparza, but only in HRD patients.In addition, for platinum-resistant ovarian cancer (PROC) patients with frequent relapse, she recommended expanding access to antibody-drug conjugate (ADC) therapies through reimbursement. Elahere is the first treatment in about a decade to demonstrate improvements in overall survival (OS) and progression-free survival (PFS) following bevacizumab.Professor Lee emphasized, “The combination therapy of olaparib, a PARP inhibitor, and bevacizumab extended progression-free survival by nearly 30 months in HRD-positive patients, reducing the risk of disease progression or death by 59%. Olaparib plus bevacizumab is the only combination that has improved overall survival in HRD-positive ovarian cancer.”She added, “The olaparib plus bevacizumab combination is the only Category 1 option recommended in NCCN guidelines and is recognized as standard therapy in major developed countries. However, in Korea, the treatment that has demonstrated overall survival benefits remains non-reimbursed, limiting patient access.”Professor Lee stated, “In the case of platinum-resistant ovarian cancer, Elahere has, for the first time in a long while since bevacizumab, significantly demonstrated an improvement in progression-free survival. As an ADC, it is designed to deliver cytotoxic agents directly to tumor cells, acting like a biologically guided missile.”Professor Lee proposed policy measures such as a pre-reimbursement post-evaluation system, flexible cost-effectiveness assessment, raising or applying flexible ICER thresholds, and incorporating clinical expert opinions more actively throughout the innovative new drug reimbursement process to ensure that unmet clinical needs and clinical necessity in actual treatment settings are faithfully reflected in reimbursement evaluations.She emphasized the need for rapid reimbursement of the bevacizumab + olaparib combination therapy for HRD-positive patients and the ADC drug Elahere for platinum-resistant cases.She concluded, “Ovarian cancer is often diagnosed at Stage III or IV and has frequent recurrence, making it the most lethal gynecologic cancer. Expanding access to innovative therapies through faster reimbursement and broader coverage is urgent. Given that ovarian cancer is a key priority in national coverage policy, prompt expansion of reimbursement access is essential.”